示例用法
如何使用Deeptools进行芯片序列分析
为了了解deeptools可以做什么,我们想简要介绍一下我们通常如何使用deeptools进行chip-seq分析。对于工具的更详细的示例和描述,只需遵循相应的链接即可。
备注
而一些工具,如 绘图针打印 特别是解决芯片序列问题,大多数工具广泛适用于深度测序数据,包括RNA序列。
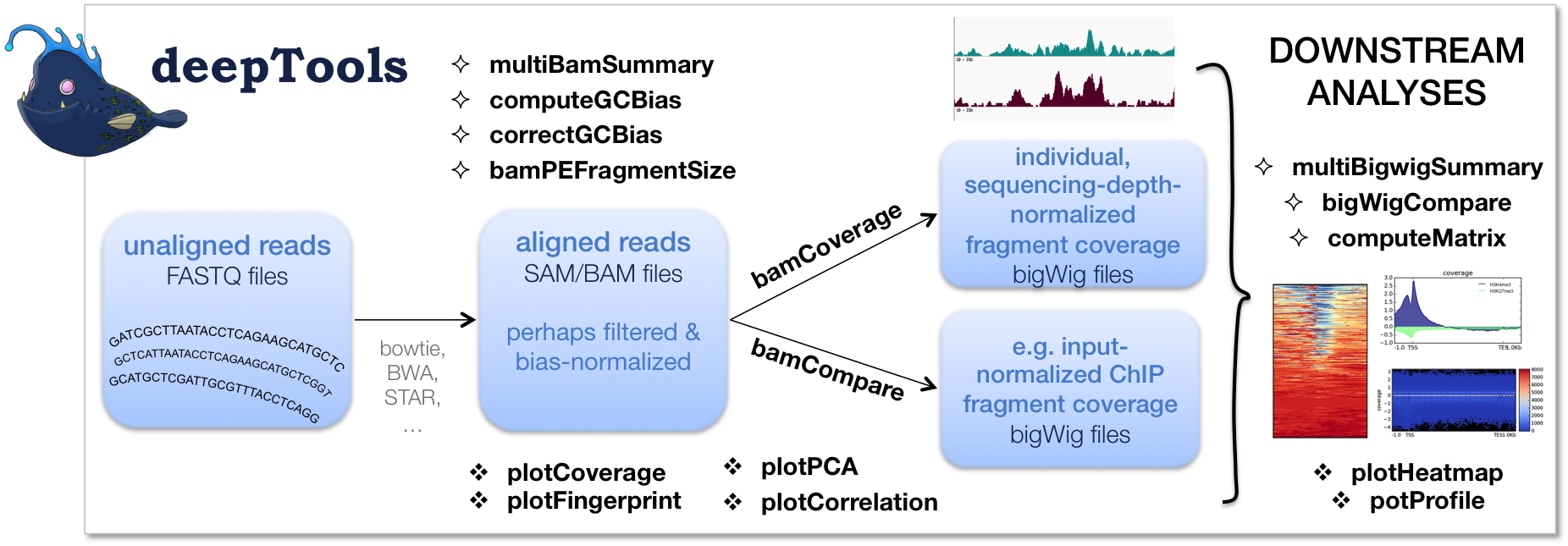
如上图所示,我们的工作通常从一个或多个 FASTQ 深度排序样本的文件。初步质量控制后使用 FASTQC ,我们将读取数据与参考基因组进行比对,例如,使用 bowtie2 . bowtie2(和其他映射工具)的标准输出是以排序和索引的形式进行的。 BAM 为所有后续deepools分析提供公共输入和起点的文件。然后,我们使用deeptools来评估对齐读取的质量:
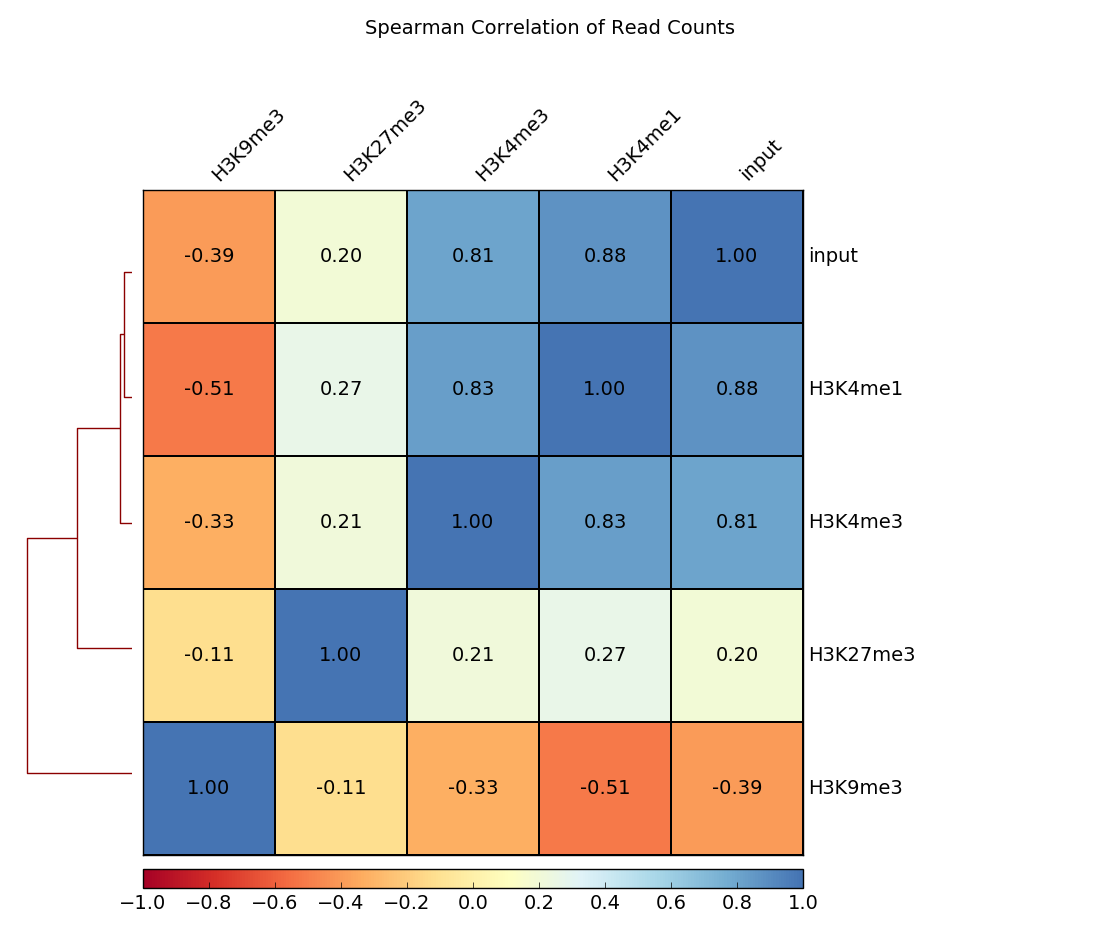
Correlation between BAM files (多BAM摘要 and 普洛相关). Together, these two modules perform a very basic test to see whether the sequenced and aligned reads meet your expectations. We use this check to assess reproducibility - either between replicates and/or between different experiments that might have used the same antibody or the same cell type, etc. For instance, replicates should correlate better than differently treated samples.
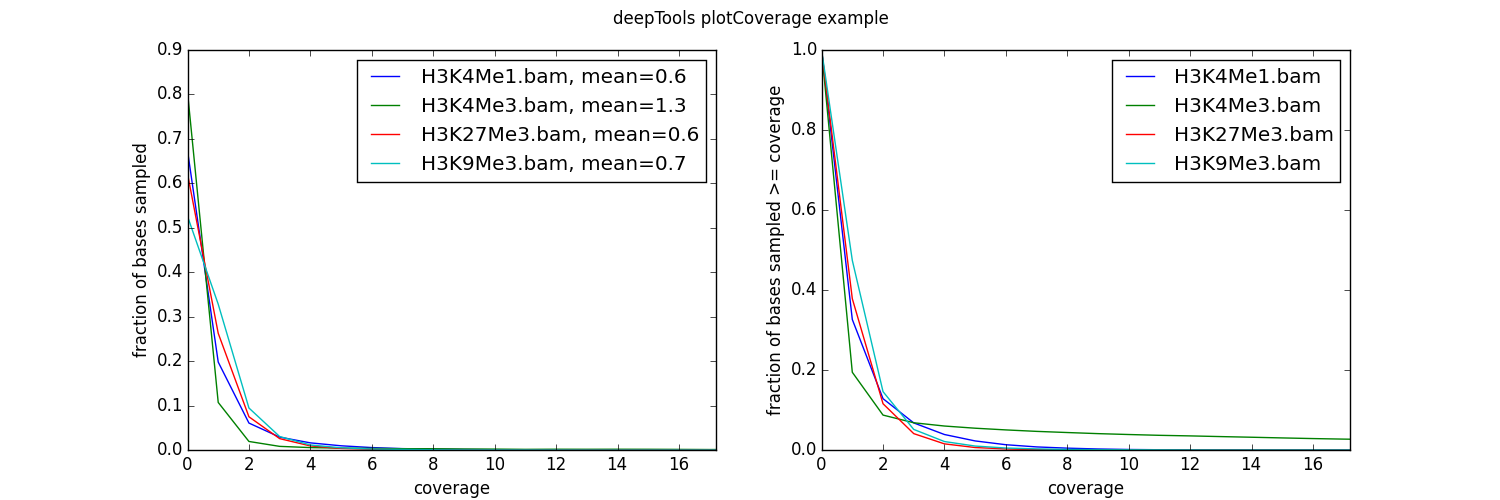
Coverage check (绘图平均值). To see how many bp in the genome are actually covered by (a good number) of sequencing reads, we use 绘图平均值 which generates two diagnostic plots that help us decide whether we need to sequence deeper or not. The option
--ignoreDuplicatesis particularly useful here!
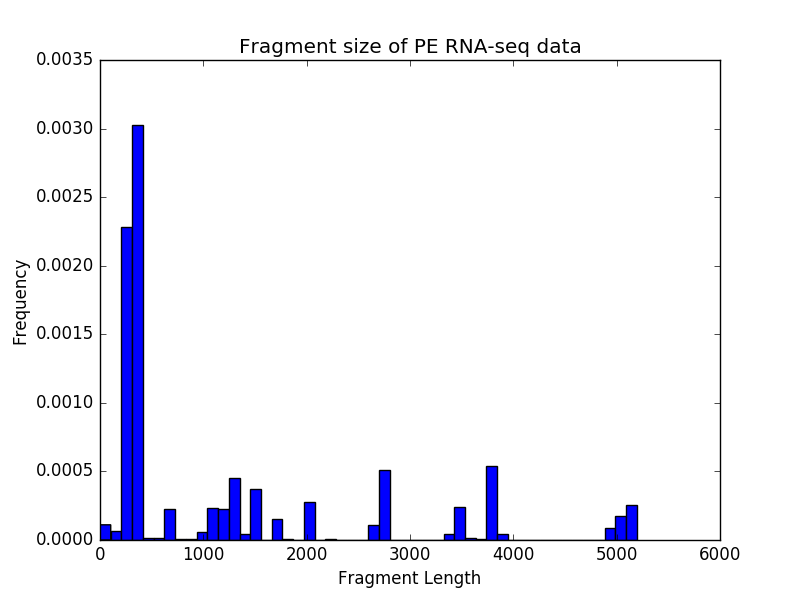
对于成对的端部样本,我们经常额外检查片段大小是否与基于库准备的预期大小差不多。模块 Bampe碎片大小 可以用来做那个。
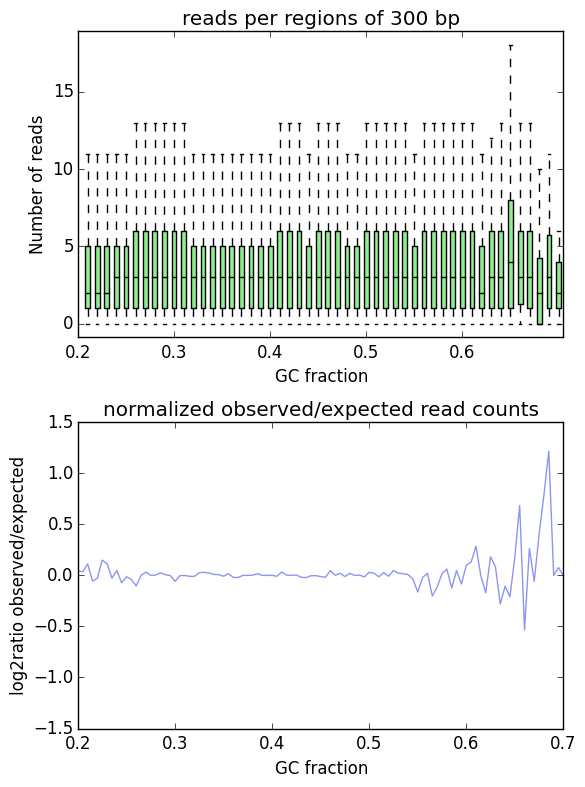
GC-bias check (计算能力). Many sequencing protocols require several rounds of PCR-based DNA amplification, which often introduces notable bias, due to many DNA polymerases preferentially amplifying GC-rich templates. Depending on the sample (preparation), the GC-bias can vary significantly and we routinely check its extent. When we need to compare files with different GC biases, we use the 纠正偏差 module. See the paper by Benjamini and Speed for many insights into this problem.
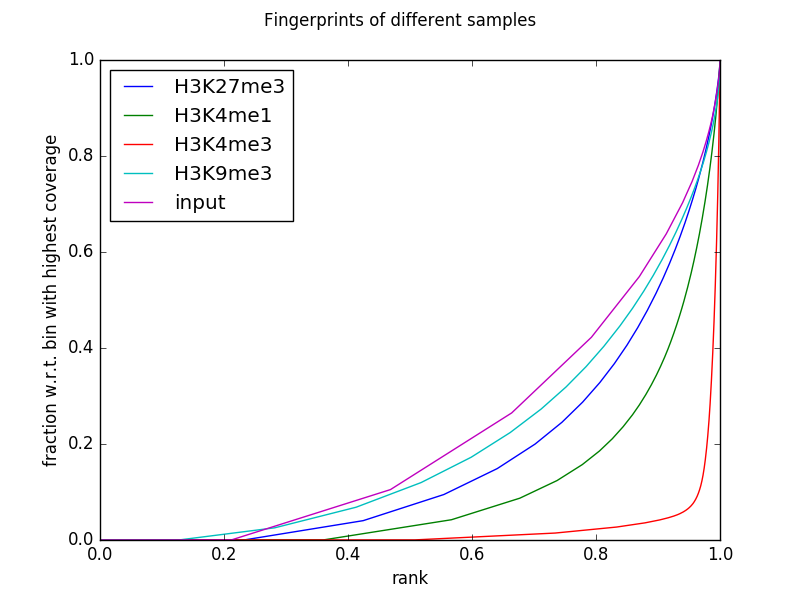
评估芯片强度 . 我们做了这个质量控制步骤,以了解芯片序列实验样本中的信噪比。它基于 Diaz et al.
一旦我们对基本质量检查满意,我们通常 转换 大 BAM 将文件转换为更精简的数据格式,通常 bigWig . 与BAM文件相比,bigwig文件有几个优势,主要是由于它们的大小显著减小:
用于数据共享和存储
基因组浏览器中的直观可视化(例如 IGV )
更有效的下游分析是可能的
Deeptools模块 BAM比较 和 BAM覆盖范围 不仅允许将BAM简单转换为bigwig(或 床面描记器 对于这一点),还要进行归一化,这样,尽管不同的样本的排序深度不同,它们还是可以进行比较。
最后,一旦所有转换的文件通过我们的目视检查(例如,使用 Integrative Genomics Viewer )下游分析的乐趣 计算机 , 地图 和 绘图配置文件 可以开始了!
deepTools Galaxy <http://deeptools.ie-freiburg.mpg.de> _. |
code @ github <https://github.com/deeptools/deepTools/> _. |