逐步协议
本节将概述如何执行许多常见任务。我们正在使用 Galaxy的截图 在这里。如果您使用的是命令行版本,那么您可以很容易地遵循给定的示例,因为绝大多数参数也会在Galaxy中显示。否则,只需键入程序名和帮助选项(例如 /deepTools/bin/bamCoverage --help ,这将显示所有可用的参数和选项。或者,您可以按照阅读docs上的工具文档的相应链接进行操作。
备注
如需支持或问题,请发帖至 Biostars . 对于错误报告和功能请求,请打开问题 <on github .
所有协议都假定您已将文件上载到安装了deeptools的Galaxy实例中,例如, deepTools Galaxy . 如果您需要帮助才能开始使用Galaxy,例如上传数据,请参见 在Galaxy中使用deepTool 和 数据导入Galaxy .
小技巧
如果您想尝试使用 样本数据 去 deepTools Galaxy -->“共享数据”-->“数据库”-->“Deeptools测试文件”。只需选择bed/bam/bigwig文件并单击“to history”。您也可以通过单击顶部的“下载”将测试数据集下载到您的计算机。
质量控制和数据处理
我已经下载/接收了一个BAM文件-如何生成一个我可以在基因组浏览器中查看的文件?
当然,你也可以在基因组浏览器中查看你的BAM文件。但是,生成一个读覆盖率较大的文件将大大减小文件的大小,它还允许您将覆盖率规范化为1X序列深度,这使得对多个文件进行可视化比较更加可行。

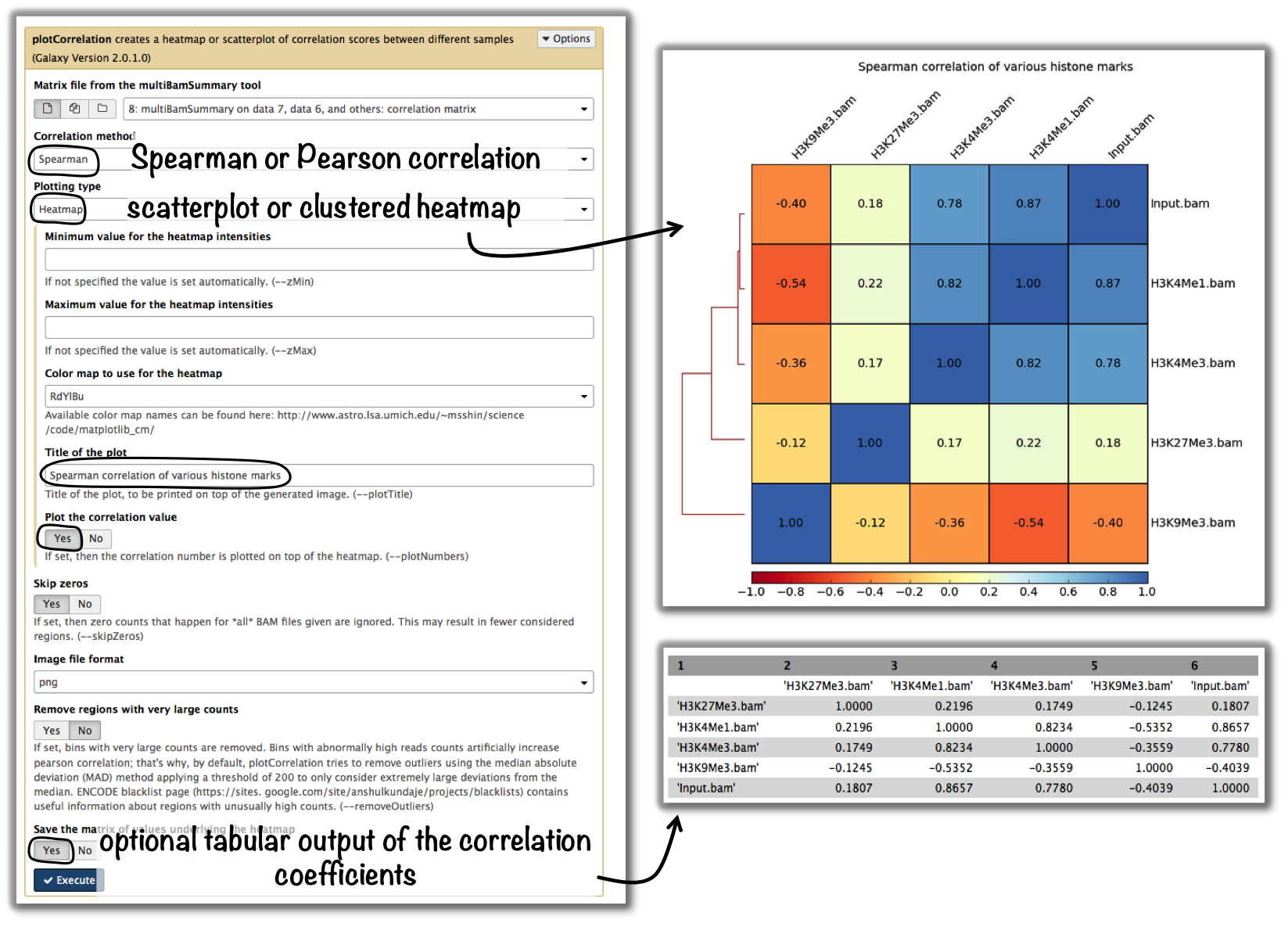
我如何评估我的序列复制的再现性?
通常,您会对不同副本和不同样本的读取覆盖率的相关性感兴趣。你想看到的是,复制应该比不复制更好地相互关联。这个 ENCODE consortium recommends 那个 [for messenger RNA, (...) biological replicates [should] display 0.9 correlation for transcripts/features] . 有关相关性计算的详细信息,请参见 普洛相关 .
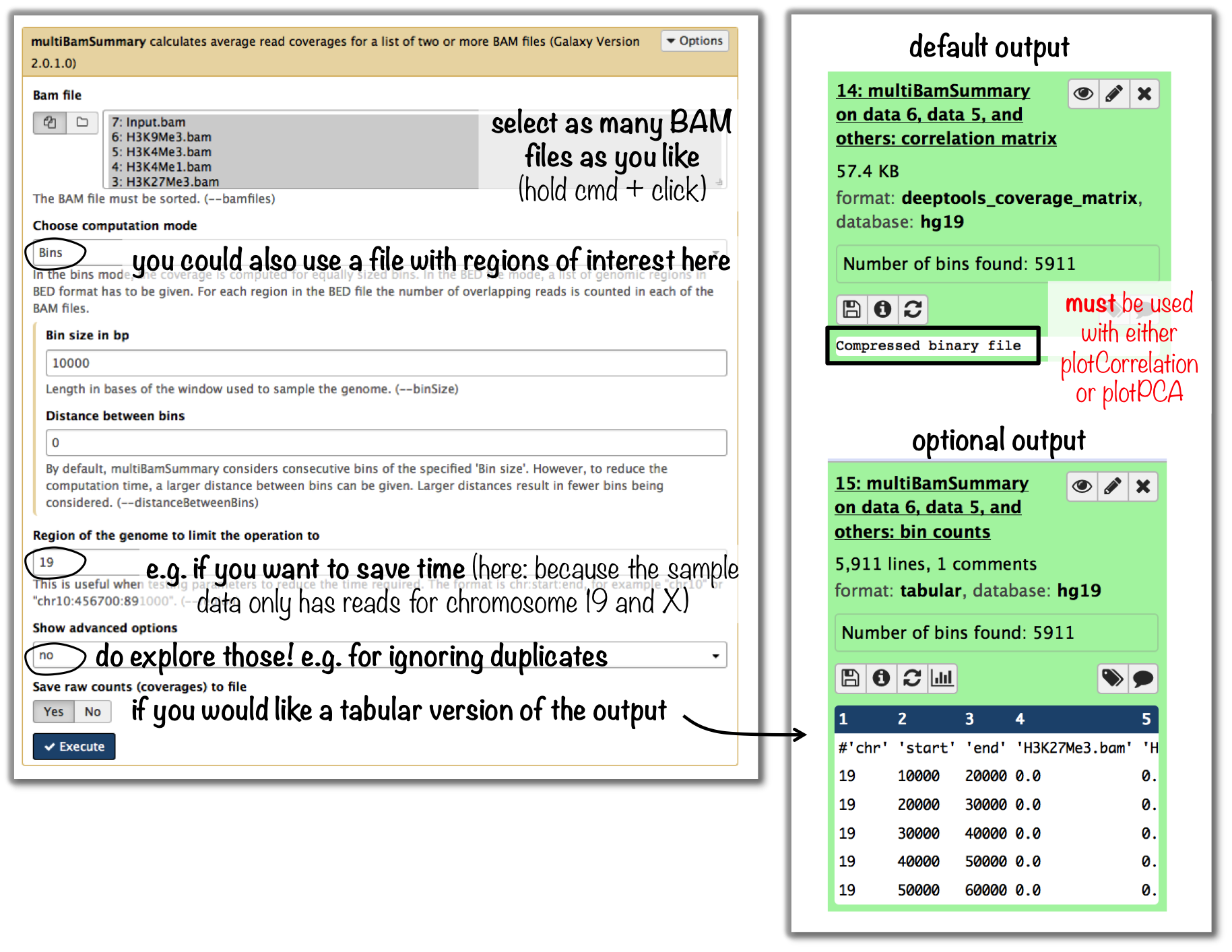
小技巧
如果要基于bigwig文件进行类似的分析,请使用该工具 multiBigwigSummary 相反。
我如何知道我的样本是否偏向GC?如果是的话,我该怎么纠正呢?
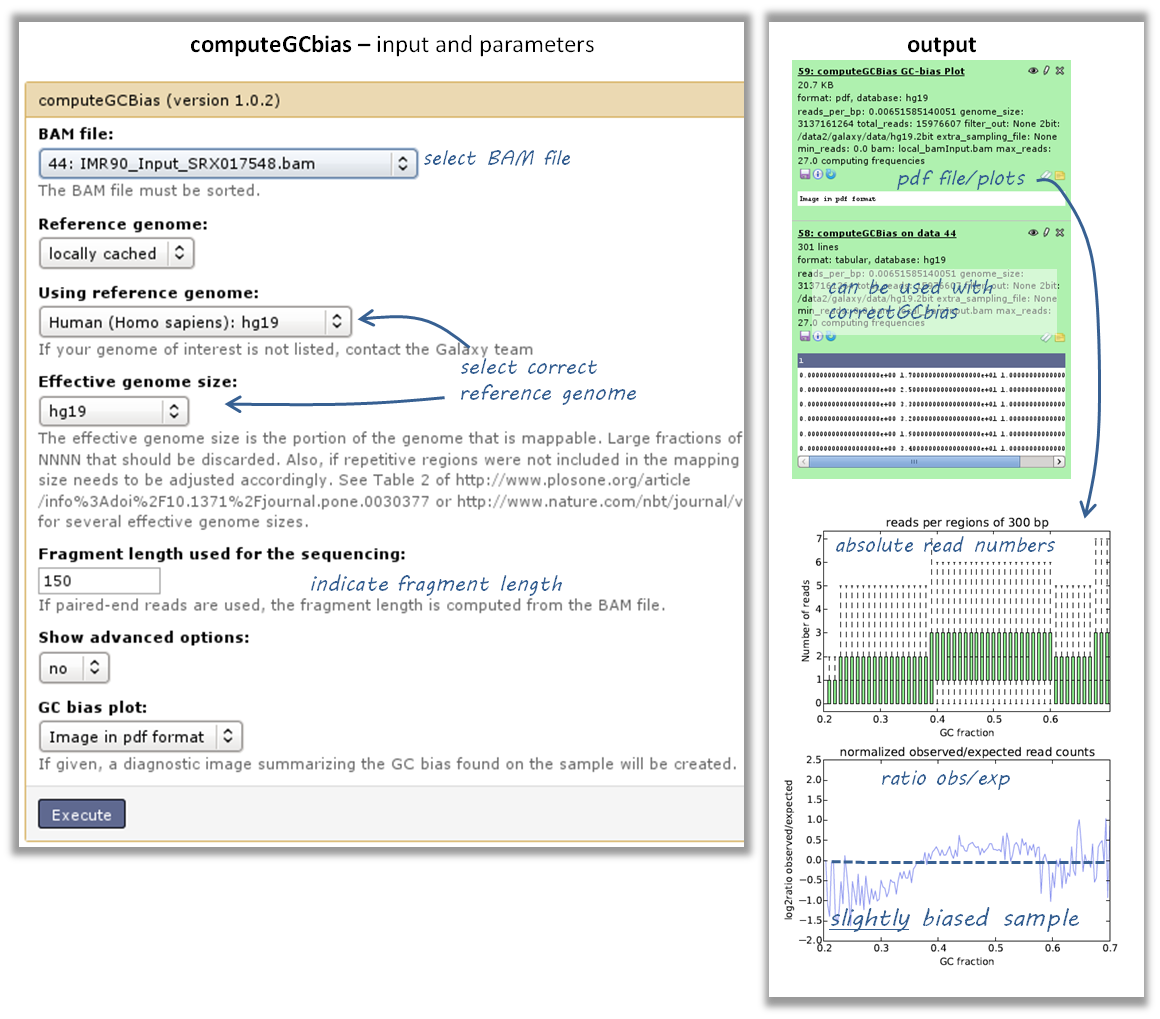
查看生成的图像并将其与示例进行比较 here
如果您的样本显示exp/obs覆盖率几乎呈线性增长(在下图的对数尺度上),那么您应该考虑纠正gc偏差。- if 您认为该数据的生物学解释可能会受到影响(例如,将其与另一个没有固有GC偏差的样品进行比较)。
GC偏差可以用工具纠正。 纠正偏差 使用必须运行的computegbias工具的第二个输出
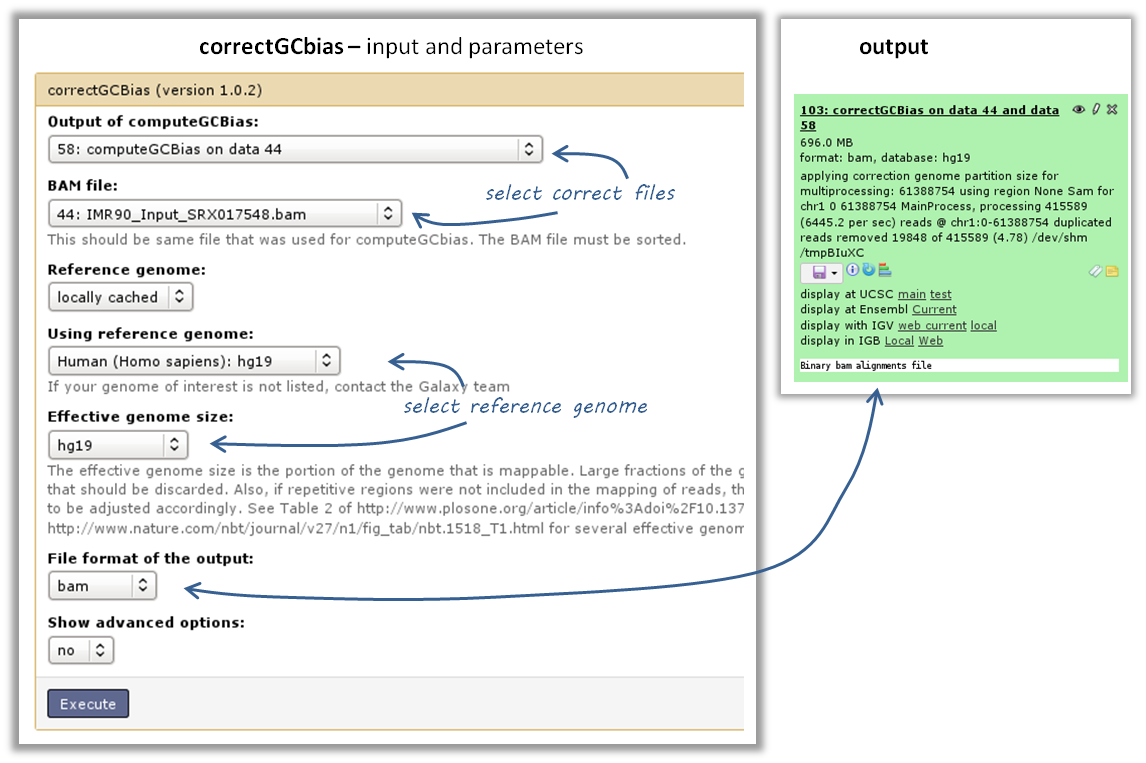
警告
correctGCbias 将向其他耗尽的区域(通常是GC不良区域)添加读取,这意味着您应该 not 根据GC更正的BAM文件删除任何下游分析中的重复项。因此,我们建议在进行更正之前删除重复项,以便只保留由GC更正过程生成的那些重复读取。
如何获得输入规范化的chip seq覆盖文件?
输入:您需要两个BAM文件,一个用于输入,一个用于chip seq实验
工具: BAM比较 芯片=处理,输入=控制样本

如何比较不同芯片实验的芯片强度?
工具: 绘图针打印
输入:要比较的任意多个chip seq样本的BAM文件(包含输入控件有助于查看希望的非强化样本是什么样子的)
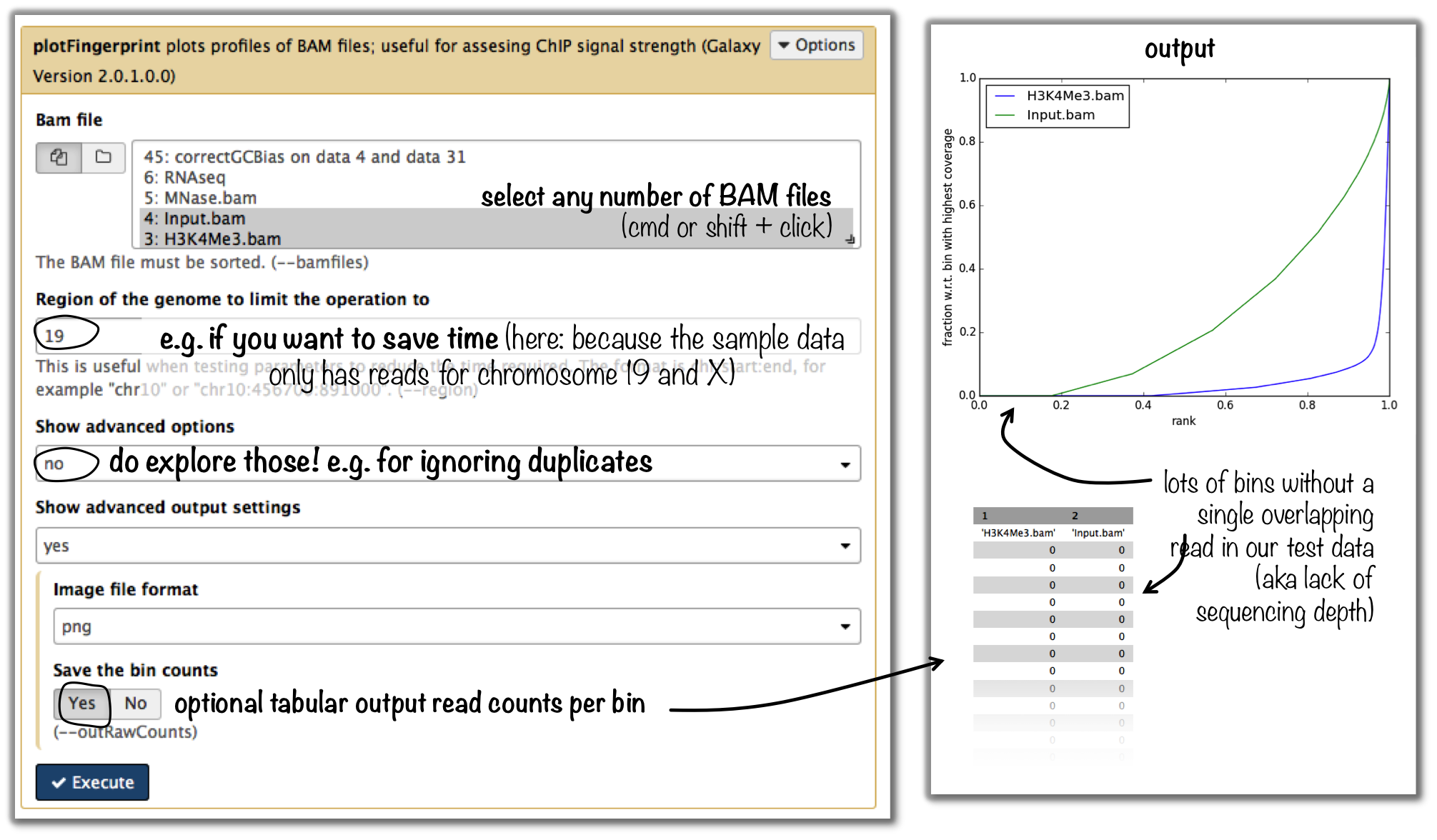
小技巧
有关绘图解释的更多详细信息,请参见 绘图针打印 或者选择Deeptools星系中的工具并向下滚动以获取更多信息。
热图和汇总图
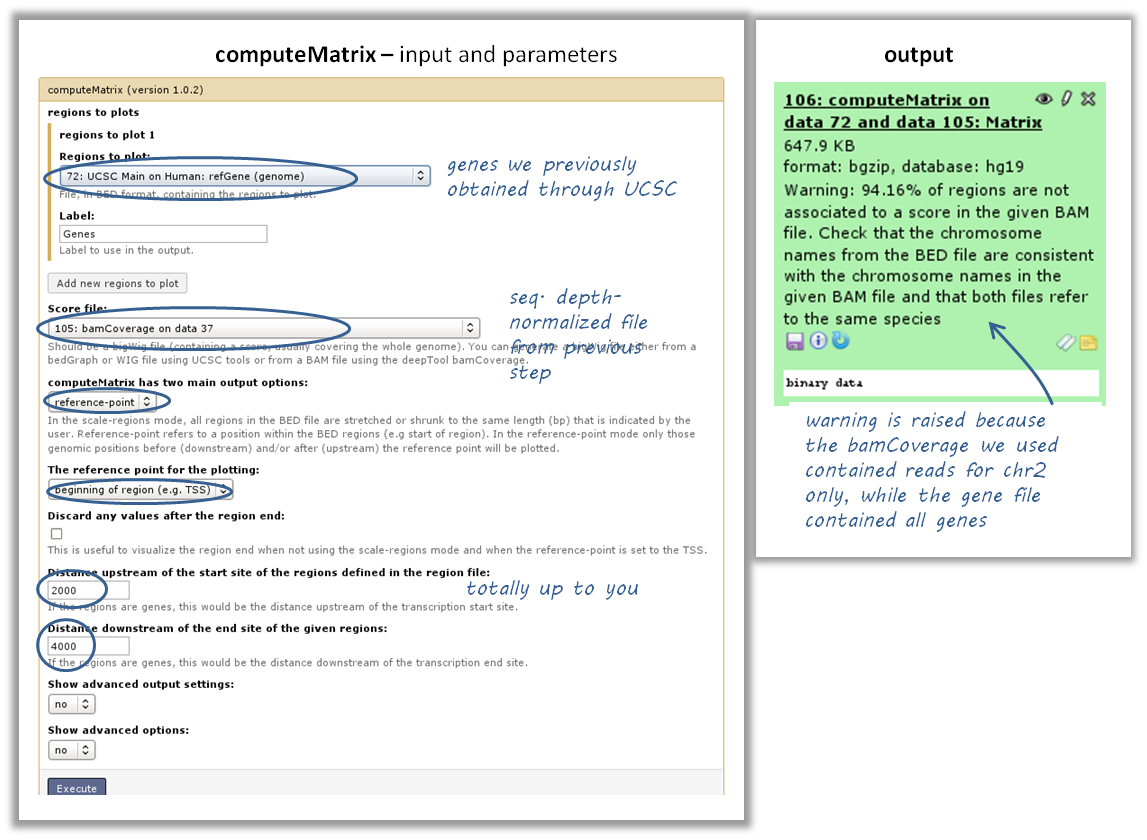
如何获得(聚集的)序列深度热图?所有基因转录起始位点周围的标准化读取覆盖率?
使用 计算机 用大人物档案和床上档案
表明
reference-point(无论您想调优其他选项,请参见下面的屏幕截图)

如何比较2个或更多不同测序实验中X特异基因和常染色体基因的平均信号?
在使用此协议之前,请确保您熟悉ComputeMatrix和PlotProfile。
- 工具:
使用简单表达式筛选任何列上的数据
计算机
绘图配置文件
(绘制多个样本的汇总图)
- 输入:
几个bigwig文件(您要比较的每个测序实验一个)
两个床文件,一个带有X染色体,一个带有常染色体基因
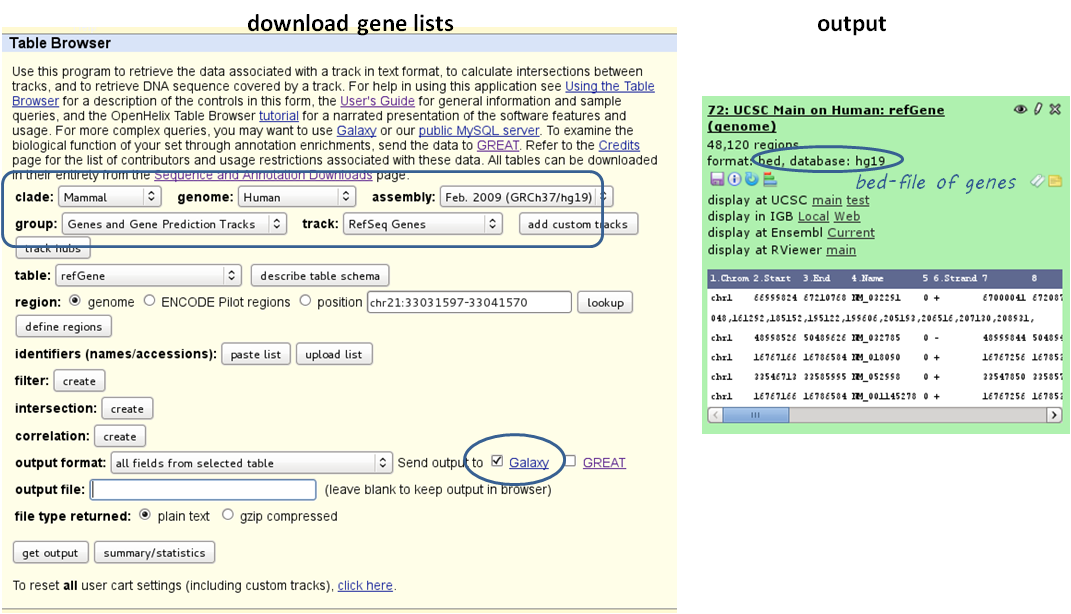
如何获取每个X染色体和常染色体基因的床文件
通过“获取数据”-->“UCSC主表浏览器”-->组:“基因和基因预测”-->跟踪(例如,“refseqgenes”-->发送到Galaxy)下载完整的基因列表
使用工具筛选列表两次 “使用简单表达式筛选任何列上的数据”
首先使用表达式:c1==“chrx”过滤所有基因的列表——>这将生成一个X连锁基因的列表。
然后重新运行过滤,现在使用c1!=“chrx”,它将生成一个不属于X染色体的基因列表(!=表示“不匹配”)
计算X和常染色体基因的平均值
使用 计算机 对于所有信号文件(bigwig格式)
提供两个过滤床文件(单击一次“添加要绘制的新区域”)并标记它们
指示相应的信号文件
现在使用 绘图配置文件 在结果文件上
重要提示:显示“高级输出选项”并选择“保存平均配置文件下的数据”-->这将生成一个表以及摘要绘图图像

deepTools Galaxy <http://deeptools.ie-freiburg.mpg.de> _. |
code @ github <https://github.com/deeptools/deepTools/> _. |