BAM覆盖范围
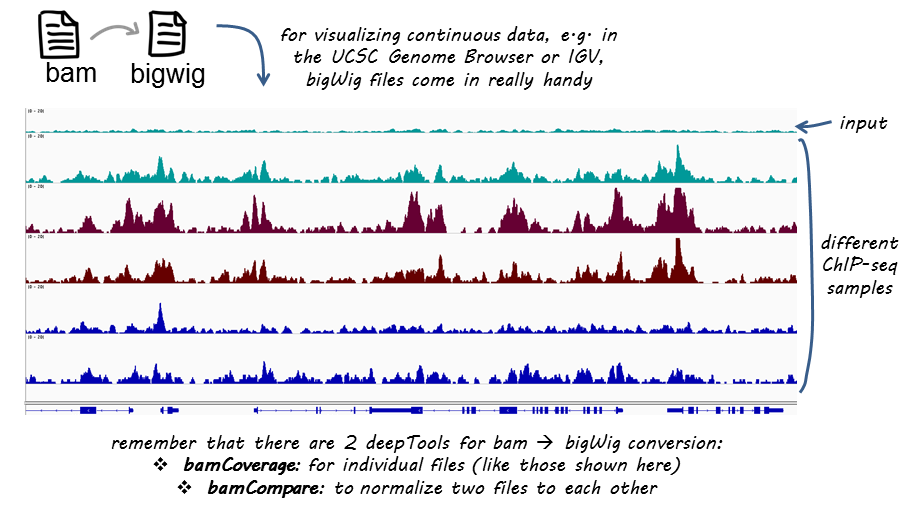
如果您不熟悉BAM、bedgraph和bigwig格式,可以在我们的 NGS术语表
This tool takes an alignment of reads or fragments as input (BAM file) and generates a coverage track (bigWig or bedGraph) as output. The coverage is calculated as the number of reads per bin, where bins are short consecutive counting windows of a defined size. It is possible to extended the length of the reads to better reflect the actual fragment length. bamCoverage offers normalization by scaling factor, Reads Per Kilobase per Million mapped reads (RPKM), counts per million (CPM), bins per million mapped reads (BPM) and 1x depth (reads per genome coverage, RPGC).
usage: bamCoverage -b reads.bam -o coverage.bw
help: bamCoverage -h / bamCoverage --help
Required arguments
- --bam, -b
BAM file to process
Output
- --outFileName, -o
Output file name.
- --outFileFormat, -of
Possible choices: bigwig, bedgraph
Output file type. Either "bigwig" or "bedgraph".
Optional arguments
- --scaleFactor
The computed scaling factor (or 1, if not applicable) will be multiplied by this. (Default: 1.0)
- --MNase
Determine nucleosome positions from MNase-seq data. Only 3 nucleotides at the center of each fragment are counted. The fragment ends are defined by the two mate reads. Only fragment lengthsbetween 130 - 200 bp are considered to avoid dinucleosomes or other artifacts. By default, any fragments smaller or larger than this are ignored. To over-ride this, use the --minFragmentLength and --maxFragmentLength options, which will default to 130 and 200 if not otherwise specified in the presence of --MNase. NOTE: Requires paired-end data. A bin size of 1 is recommended.
- --Offset
Uses this offset inside of each read as the signal. This is useful in cases like RiboSeq or GROseq, where the signal is 12, 15 or 0 bases past the start of the read. This can be paired with the --filterRNAstrand option. Note that negative values indicate offsets from the end of each read. A value of 1 indicates the first base of the alignment (taking alignment orientation into account). Likewise, a value of -1 is the last base of the alignment. An offset of 0 is not permitted. If two values are specified, then they will be used to specify a range of positions. Note that specifying something like --Offset 5 -1 will result in the 5th through last position being used, which is equivalent to trimming 4 bases from the 5-prime end of alignments. Note that if you specify --centerReads, the centering will be performed before the offset.
- --filterRNAstrand
Possible choices: forward, reverse
Selects RNA-seq reads (single-end or paired-end) originating from genes on the given strand. This option assumes a standard dUTP-based library preparation (that is, --filterRNAstrand=forward keeps minus-strand reads, which originally came from genes on the forward strand using a dUTP-based method). Consider using --samExcludeFlag instead for filtering by strand in other contexts.
- --version
show program's version number and exit
- --binSize, -bs
Size of the bins, in bases, for the output of the bigwig/bedgraph file. (Default: 50)
- --region, -r
Region of the genome to limit the operation to - this is useful when testing parameters to reduce the computing time. The format is chr:start:end, for example --region chr10 or --region chr10:456700:891000.
- --blackListFileName, -bl
A BED or GTF file containing regions that should be excluded from all analyses. Currently this works by rejecting genomic chunks that happen to overlap an entry. Consequently, for BAM files, if a read partially overlaps a blacklisted region or a fragment spans over it, then the read/fragment might still be considered. Please note that you should adjust the effective genome size, if relevant.
- --numberOfProcessors, -p
Number of processors to use. Type "max/2" to use half the maximum number of processors or "max" to use all available processors. (Default: 1)
- --verbose, -v
Set to see processing messages.
Read coverage normalization options
- --effectiveGenomeSize
The effective genome size is the portion of the genome that is mappable. Large fractions of the genome are stretches of NNNN that should be discarded. Also, if repetitive regions were not included in the mapping of reads, the effective genome size needs to be adjusted accordingly. A table of values is available here: http://deeptools.readthedocs.io/en/latest/content/feature/effectiveGenomeSize.html .
- --normalizeUsing
Possible choices: RPKM, CPM, BPM, RPGC, None
Use one of the entered methods to normalize the number of reads per bin. By default, no normalization is performed. RPKM = Reads Per Kilobase per Million mapped reads; CPM = Counts Per Million mapped reads, same as CPM in RNA-seq; BPM = Bins Per Million mapped reads, same as TPM in RNA-seq; RPGC = reads per genomic content (1x normalization); Mapped reads are considered after blacklist filtering (if applied). RPKM (per bin) = number of reads per bin / (number of mapped reads (in millions) * bin length (kb)). CPM (per bin) = number of reads per bin / number of mapped reads (in millions). BPM (per bin) = number of reads per bin / sum of all reads per bin (in millions). RPGC (per bin) = number of reads per bin / scaling factor for 1x average coverage. None = the default and equivalent to not setting this option at all. This scaling factor, in turn, is determined from the sequencing depth: (total number of mapped reads * fragment length) / effective genome size. The scaling factor used is the inverse of the sequencing depth computed for the sample to match the 1x coverage. This option requires --effectiveGenomeSize. Each read is considered independently, if you want to only count one mate from a pair in paired-end data, then use the --samFlagInclude/--samFlagExclude options. (Default: None)
- --exactScaling
Instead of computing scaling factors based on a sampling of the reads, process all of the reads to determine the exact number that will be used in the output. This requires significantly more time to compute, but will produce more accurate scaling factors in cases where alignments that are being filtered are rare and lumped together. In other words, this is only needed when region-based sampling is expected to produce incorrect results.
- --ignoreForNormalization, -ignore
A list of space-delimited chromosome names containing those chromosomes that should be excluded for computing the normalization. This is useful when considering samples with unequal coverage across chromosomes, like male samples. An usage examples is --ignoreForNormalization chrX chrM.
- --skipNonCoveredRegions, --skipNAs
This parameter determines if non-covered regions (regions without overlapping reads) in a BAM file should be skipped. The default is to treat those regions as having a value of zero. The decision to skip non-covered regions depends on the interpretation of the data. Non-covered regions may represent, for example, repetitive regions that should be skipped.
- --smoothLength
The smooth length defines a window, larger than the binSize, to average the number of reads. For example, if the --binSize is set to 20 and the --smoothLength is set to 60, then, for each bin, the average of the bin and its left and right neighbors is considered. Any value smaller than --binSize will be ignored and no smoothing will be applied.
Read processing options
- --extendReads, -e
This parameter allows the extension of reads to fragment size. If set, each read is extended, without exception. NOTE: This feature is generally NOT recommended for spliced-read data, such as RNA-seq, as it would extend reads over skipped regions. Single-end: Requires a user specified value for the final fragment length. Reads that already exceed this fragment length will not be extended. Paired-end: Reads with mates are always extended to match the fragment size defined by the two read mates. Unmated reads, mate reads that map too far apart (>4x fragment length) or even map to different chromosomes are treated like single-end reads. The input of a fragment length value is optional. If no value is specified, it is estimated from the data (mean of the fragment size of all mate reads).
- --ignoreDuplicates
If set, reads that have the same orientation and start position will be considered only once. If reads are paired, the mate's position also has to coincide to ignore a read.
- --minMappingQuality
If set, only reads that have a mapping quality score of at least this are considered.
- --centerReads
By adding this option, reads are centered with respect to the fragment length. For paired-end data, the read is centered at the fragment length defined by the two ends of the fragment. For single-end data, the given fragment length is used. This option is useful to get a sharper signal around enriched regions.
- --samFlagInclude
Include reads based on the SAM flag. For example, to get only reads that are the first mate, use a flag of 64. This is useful to count properly paired reads only once, as otherwise the second mate will be also considered for the coverage. (Default: None)
- --samFlagExclude
Exclude reads based on the SAM flag. For example, to get only reads that map to the forward strand, use --samFlagExclude 16, where 16 is the SAM flag for reads that map to the reverse strand. (Default: None)
- --minFragmentLength
The minimum fragment length needed for read/pair inclusion. This option is primarily useful in ATACseq experiments, for filtering mono- or di-nucleosome fragments. (Default: 0)
- --maxFragmentLength
The maximum fragment length needed for read/pair inclusion. (Default: 0)
使用提示
较小的bin大小值将导致覆盖范围跟踪的更高分辨率,但也会导致更大的文件大小。
1X规范化(RPGC)要求输入 有效基因组大小 这是参考基因组的可映射部分。当然,这个值是特定于物种的。这个工具的命令行帮助为许多模型物种提供了建议。
对于某些研究来说,排除某些染色体可能是有用的,因为雄性小鼠含有一对常染色体,但通常只有一条X染色体。
默认情况下,读取长度为 NOT 扩展!这是首选的设置 spliced-read 像RNA序列这样的数据,人们通常只想依赖于检测到的读取位置。读取扩展将忽略片段未映射部分中的潜在拼接站点。其他数据,如chip seq,其中片段已知是连续映射的,应使用读取扩展处理。 (
--extendReads [INTEGER])对于成对的结束数据,片段长度通常由两个读取匹配项定义。用户提供的片段长度仅用作单子或配偶读取距离太远(距离大于片段长度的四倍或位于不同染色体上)的映射的回退。
警告
如果您已经使用 correctGCbias 你绝对应该 NOT 设置参数 --ignoreDuplicates 你说什么?
备注
和BAM文件一样,bigwig文件是压缩的二进制文件。如果您想查看覆盖率值,请选择床位图输出方式 --outFileFormat .
chip seq的用法示例
这是使用其他选项的Chip Seq数据的示例(更小的bin大小以获得更高的分辨率,将覆盖范围规范化为1X小鼠基因组大小,在规范化步骤中不包括染色体x,并扩展读取):
bamCoverage --bam a.bam -o a.SeqDepthNorm.bw \
--binSize 10
--normalizeUsing RPGC
--effectiveGenomeSize 2150570000
--ignoreForNormalization chrX
--extendReads
如果你用 --outFileFormat bedgraph ,您可以很容易地到达结果文件的峰值。
$ head SeqDepthNorm_chr19.bedgraph
19 60150 60250 9.32
19 60250 60450 18.65
19 60450 60650 27.97
19 60650 60950 37.29
19 60950 61000 27.97
19 61000 61050 18.65
19 61050 61150 27.97
19 61150 61200 18.65
19 61200 61300 9.32
19 61300 61350 18.65
如您所见,每行对应一个区域。如果连续的容器具有相同的读取重叠数,则它们将被合并。
RNA序列的用法示例
请注意,某些BAM文件是基于SAM标志筛选的 (Explain SAM flags )
普通重量级音轨
bamCoverage -b a.bam -o a.bw
每条钢绞线的单独轨道
有时产生两个独立的 大人物 分别读取正向和反向链上的所有文件。从Deeptools 2.2版开始,可以简单地使用 --filterRNAstrand 选项,例如 --filterRNAstrand forward 或 --filterRNAstrand reverse . 它处理成对的端数据集和单端数据集。有关Deeptools的旧版本,请参阅下面的说明。
备注
这个 --filterRNAstrand 选项假定由Illumina dutp/nsr/nnsr方法生成的序列库,这是最常用的库准备方法,其中read 2(r2)是RNA链的方向。( reverse-stranded 类库)。然而,也存在其他方法,它们在RNA链方向上产生read r1。 (see this review )对于这些类库, --filterRNAstrand 会有相反的行为,即 --filterRNAstrand forward 会给你反向链信号,反之亦然。
2.2之前的版本
为了遵循这些例子,你需要知道 -f 会告诉你的 samtools view 到 包括 使用指示标志读取,而 -F 会导致 排除 用各自的标志读取。
For a stranded `single-end` library
# Forward strand
bamCoverage -b a.bam -o a.fwd.bw --samFlagExclude 16
# Reverse strand
bamCoverage -b a.bam -o a.rev.bw --samFlagInclude 16
For a stranded `paired-end` library
现在,这变得有点麻烦,但是Deeptools的未来版本将使这更直接。现在,请忍受我们的压力,也许可以读一些山姆的旗帜,例如 here .
对于成对的端部样本,我们假设一对合适的对应该在相反的线上有配偶,其中Illumina线串特定协议产生读取 R2-R1 方向。我们基本上按照给定的配方 in this biostars tutorial .
获取源于 前股 :
# include reads that are 2nd in a pair (128);
# exclude reads that are mapped to the reverse strand (16)
$ samtools view -b -f 128 -F 16 a.bam > a.fwd1.bam
# exclude reads that are mapped to the reverse strand (16) and
# first in a pair (64): 64 + 16 = 80
$ samtools view -b -f 80 a.bam > a.fwd2.bam
# combine the temporary files
$ samtools merge -f fwd.bam a.fwd1.bam a.fwd2.bam
# index the filtered BAM file
$ samtools index fwd.bam
# run bamCoverage
$ bamCoverage -b fwd.bam -o a.fwd.bigWig
# remove the temporary files
$ rm a.fwd*.bam
获取源于 反向股 :
# include reads that map to the reverse strand (128)
# and are second in a pair (16): 128 + 16 = 144
$ samtools view -b -f 144 a.bam > a.rev1.bam
# include reads that are first in a pair (64), but
# exclude those ones that map to the reverse strand (16)
$ samtools view -b -f 64 -F 16 a.bam > a.rev2.bam
# merge the temporary files
$ samtools merge -f rev.bam a.rev1.bam a.rev2.bam
# index the merged, filtered BAM file
$ samtools index rev.bam
# run bamCoverage
$ bamCoverage -b rev.bam -o a.rev.bw
# remove temporary files
$ rm a.rev*.bam
deepTools Galaxy <http://deeptools.ie-freiburg.mpg.de> _. |
code @ github <https://github.com/deeptools/deepTools/> _. |