一般常见问题
小技巧
如需支持或问题,请发帖至 Biostars . 对于错误报告和功能请求,请打开问题 <on github .
备注
我们还有一个 Galaxy相关常见问题 有关于星系而不是deepTool的问题。
Deeptools如何处理来自配对端序列的数据?
一般来说,所有模块都在工作 BAM 文件夹 (multiBamSummary , bamCoverage , bamCompare , plotFingerprint , computeGCBias )自动识别成对的结束序列数据,并根据读取对之间的距离使用片段大小。您可以通过选项绕过配对上的典型片段处理 --doNotExtendPairedEnds (可在Galaxy的“高级选项”下找到)。
如何在计算时间很短的情况下测试工具?
当你在玩弄这些工具以了解它们将产生什么样的结果时,你可以将操作限制在一个染色体或一个特定的区域以节省时间。在Galaxy中,您可以在“高级输出选项”-->“限制操作的基因组区域”下找到这一点。调用命令行选项 --region (CHR:START:END) .
以下工具当前具有此选项:
它的工作原理如下:首先, 整个的 基因组在 BAM 将对文件进行审查和抽样, then 所有不与用户指定区域重叠的区域或采样箱都将被丢弃。
备注
您只能将操作限制为 one 染色体(或 one 染色体上的特定位点)。如果要将操作限制到多个区域,请参阅下一个问题的答案。
我可以指定一个以上的染色体在 --regions 选择?
简短的回答是:不。
一些程序允许指定特定区域。对于这些,输入的格式必须为 chr:start:end 例如“chr10”或“chr10:456700:891000”。
对于这些程序来说, 不可能 表示多个区域,例如chr10、chr11- 这个不行 !以下是一些解决方法的建议,如果您不需要这样做:
一般解决方法
因为所有的工具都有 --region 选项工作 BAM 文件,你可以 过滤你的阅读 在运行程序之前,例如使用 intersectBed 具有 --abam 或 samtools view . 然后将生成的(较小的)BAM文件与您选择的deeptools程序一起使用。
$ samtools view -b -L regionsOfInterest.bed Reads.bam > ReadsOverlappingWithRegionsOfInterest.bam
或
$ intersectBed -abam Reads.bam -b regionsOfInterest.bed > ReadsOverlappingWithRegionsOfInterest.bam
内置解决方案
computeGCBias 和 multiBamSummary 提供内置解决方案,这样您就不需要求助于Deeptools之外的工具。
我应该什么时候排除地区 computeGCBias 是吗?
备注
一般来说,我们建议仅纠正GC偏差(使用 计算能力 然后 纠正偏差 )如果大部分基因组(例如,对于小鼠和人类基因组,30-60%之间的区域)是GC偏向的。 and 您希望将此示例与另一个不偏向GC的示例进行比较。
有时,预期存在一定的GC偏差,例如哺乳动物样品中H3K4me3的芯片样品,其中预期富含GC的启动子会被富集。为了不混淆由库准备引起的GC偏差和固有的、预期的GC偏差,我们将提供一个区域文件的可能性整合到 computeGCBias 这将从GC偏差计算中排除。这个文件通常应该包含那些我们期望显著丰富的区域。这使得工具可以集中于背景区域。
我什么时候用 bamCoverage 或 bamCompare 是吗?
两种工具都产生 大人物 文件,即它们从 BAM 把一个固定大小的基因组区域的分数归档。唯一的区别是 BAM文件数 工具用作输入:while BAM覆盖范围 将只获取一个BAM文件并生成一个覆盖率文件,该文件主要针对排序深度进行规范化, BAM比较 将采取 two BAM 可以使用几个数学运算相互比较的文件。
bamCompare 将始终对排序深度进行规范化,例如 bamCoverage ,但它将根据用户选择的内容执行其他计算,例如:
在处理RNA序列数据时,我应该注意什么?
默认情况下,deeptools( 从版本2开始 )利用对齐文件的所谓雪茄串中存储的信息 (SAM/BAM specification )雪茄精确地告诉读者参考图的哪个基准点,相应地,在跨越引言的阅读中跳过哪个基准点。这些所谓的拆分读取由Deeptools2.0的所有模块本地处理。
警告
一般来说 not 建议激活deeptools参数 --extendReads 对于RNA序列数据。
这是因为在实际读取序列之外没有关于片段对齐的已验证信息。一个简单的扩展读取未覆盖的部分对于很多片段来说可能是错误的!同时激活读取扩展 停用 雪茄的使用。
Computematrix如何处理重叠的基因组区域?
如果 BED 文件提供给 计算机 包含重叠的区域,但将按原样处理。如果您想防止这种情况发生,请清洁 BED 使用前的文件 computeMatrix . 有几种方法可以修改您的床位文件。
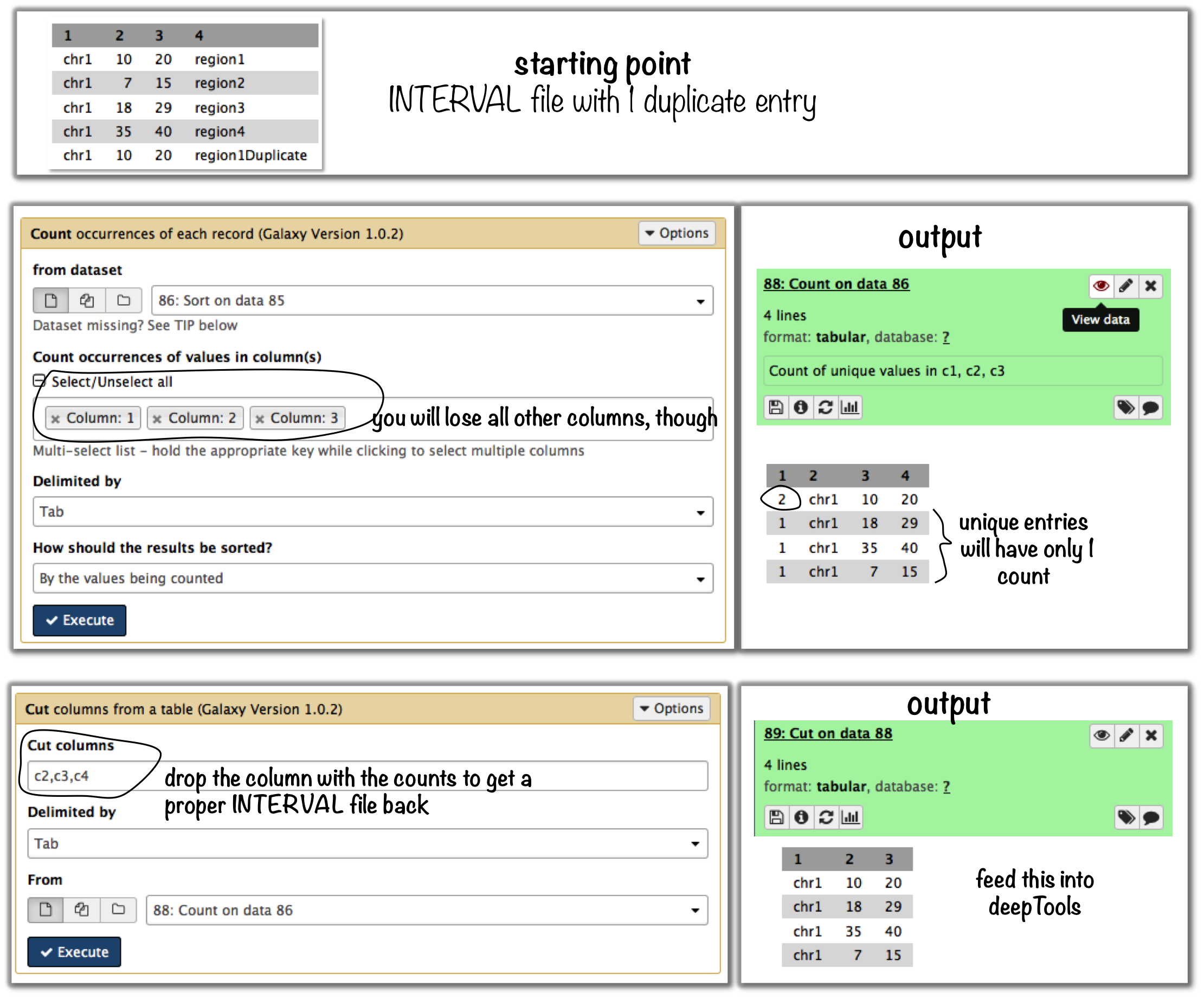
假设您的文件如下:
$ cat testBed.bed
chr1 10 20 region1
chr1 7 15 region2
chr1 18 29 region3
chr1 35 40 region4
chr1 10 20 region1Duplicate
基于Galaxy的工作
删除带有 完全相同的 基因组坐标,首先使用工具“计数”,然后过滤掉所有出现多次的条目。
基于命令行的工作区
如果你只是想消除 完全相同的 条目(此处:区域1和区域1副本),使用
sort和uniq在shell中(请注意,相同区域的标签是不同的-asuniq只能忽略文件开头的字段,使用rev要还原已排序的文件,则uniq忽略第一个字段(现在是名称列),然后返回:$ sort -k1,1 -k2,2n testBed.bed | rev | uniq -f1 | rev chr1 10 20 region1 chr1 7 15 region2 chr1 18 29 region3 chr1 35 40 region4
如果你愿意的话 合并所有重叠区域 变成一个大的,用
mergeBed从Bedtools套房:同样,必须首先对床位文件进行排序
-n和-nms告诉mergeBed输出重叠区域的数目及其名称在生成的文件中,区域1、2和3合并为:
$ sort -k1,1 -k2,2n testBed.bed | mergeBed -i stdin -n -nms chr1 7 29 region2;region1;region1Duplicate;region3 4 chr1 35 40 region4 1
如果你愿意的话 仅保留不重叠的区域 对于同一床文件中的任何其他区域,请使用相同的
mergeBed常规,但随后过滤掉合并了多个区域的区域。这个
awk命令将检查每行的最后一个字段 ($NF)并将打印原始行 ($0)仅当最后一个字段包含小于2的数字时::$ sort -k1,1 -k2,2n testBed.bed | mergeBed -i stdin -n -nms | awk '$NF < 2 {print $0}' chr1 35 40 region4 1
为什么heatmap中的最大值不等于矩阵中的最大值?
在绘制heatmap之前,对矩阵进行额外的处理,例如离群值去除。我们发现这在大多数情况下都是有益的。您可以通过手动设置来覆盖此项 --zMax 和/或 `--zMin ,分别。
我生成的heatmap看起来非常“粗糙”,我想要更细粒度的图像。
减少 料仓尺寸 使用生成矩阵时 computeMatrix
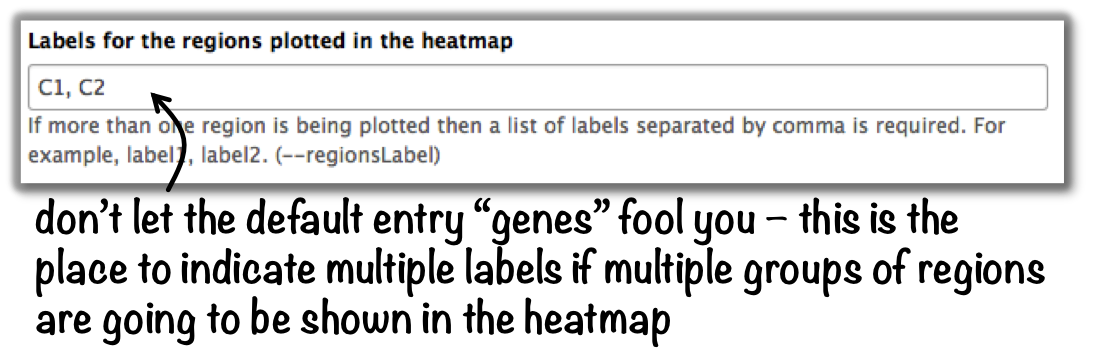
如何更改k-means集群heatmap中集群的自动标签?
每个集群的处理方式与不同的区域组完全相同。因此,可以使用相同的选项定义最终热图的标签:
- 在Galaxy:
plotaMap-->“高级输出选项”-->“热图中绘制区域的标签”。
如果为k-means clustering指定了2个集群,请在此处输入:c1、c2、->而不是完整的默认标签(“cluster 1”),heatmap将标记为缩写。
在命令行中,使用 --regionsLabel 用于定义区域的自定义名称的选项。
如何手动指定多组区域(而不是群集)?
只需指定多个床位文件(例如genes.bed、exons.bed和intron.bed)。这在Galaxy和命令行中都有效。
当我处理一个基因组草案时,我需要注意什么?
如果你的基因组不包含在我们的标准数据集中,那么你需要以下信息:
有效基因组大小 -这主要是为了 bamCoverage 和 bamCompare 见 below 详情
2bit格式的参考基因组序列 -这是为了 computeGCBias 见 2bit 详情
我如何使用除/tmp之外的临时目录?
您可以设置您的 $TMPDIR 环境变量添加到不同的目录。
我如何计算一个不在你名单上的有机体的有效基因组大小?
目前我们没有为此目的提供工具,因此您暂时必须在deeptools之外找到解决方案。
“真正的”有效基因组大小是基因组的一部分,即 唯一可映射 . 这意味着该值将取决于基因组特性(多少重复元素,组装质量等)和序列读取的长度,因为1亿36 bp读取可能覆盖不到1亿100 bp读取。
我们目前为您提供以下选项:
使用一个 GEM
使用 faCount (仅当您也允许非唯一对齐读取时!)
使用 bamCoverage
使用 GEM
有一个工具承诺计算给定读取长度(k-mer长度)的任何基因组的可映射性: GEM-Mappability Calculator . 根据此回复 here ,您可以通过计算“!”的数目来计算运行此程序后的有效基因组大小。它代表独特的可映射区域。
使用 faCount
如果您正在使用 bowtie2 ,哪个报告 多映射器 (即, non-uniquely 映射读取)作为默认设置,可以使用 从UCSC工具进行facount 报告由“n”表示的基因组装配中缺失的碱基总数和碱基数量。有效的基因组大小将是碱基对的总数减去“n”的总数。下面是一个输出示例 faCount 在 D.黑腹蛇 基因组版本dm3:
$ UCSCtools/faCount dm3.fa
#seq len A C G T N cpg
chr2L 23011544 6699731 4811687 4815192 6684734 200 926264
chr2LHet 368872 90881 58504 57899 90588 71000 10958
chr2R 21146708 6007371 4576037 4574750 5988450 100 917644
chr2RHet 3288761 828553 537840 529242 826306 566820 99227
chr3L 24543557 7113242 5153576 5141498 7135141 100 995078
chr3LHet 2555491 725986 473888 479000 737434 139183 89647
chr3R 27905053 7979156 5995211 5980227 7950459 0 1186894
chr3RHet 2517507 678829 447155 446597 691725 253201 84175
chr4 1351857 430227 238155 242039 441336 100 43274
chrU 10049037 2511952 1672330 1672987 2510979 1680789 335241
chrUextra 29004656 7732998 5109465 5084891 7614402 3462900 986216
chrX 22422827 6409325 4742952 4748415 6432035 90100 959534
chrXHet 204112 61961 40017 41813 60321 0 754
chrYHet 347038 74566 45769 47582 74889 104232 8441
chrM 19517 8152 2003 1479 7883 0 132
total 168736537 47352930 33904589 33863611 47246682 6368725 6650479
在本例中:总数bp=168736537总数n=6368725
警告
这种方法只有在多个映射器随机分配到它们可能的位置时才有效(在这种情况下,有效的基因组大小只是非N碱基的数量)。
使用 bamCoverage
如果你有一个你希望基因组被完全覆盖的样本,例如从基因组测序中,一个非常小的解决方案就是使用 BAM覆盖范围 箱子大小为1 bp --outFileFormat 选项设置为“床位图”。然后,您可以计算非零箱(碱基)的数量,这将指示此特定样本的可映射基因组大小。
使用 genomeCoverageBed
genomeCoverageBed from the BEDtools suite can be used to calculate the number of bases in the genome for which 0 overlapping reads can be found. As described on the BEDtools website (转到热那梅科夫描述),您需要:
一份包含你的样本有机体染色体大小的文件
位置排序的BAM文件
$ bedtools genomecov -ibam sortedBAMfile.bam -g genome.size
在哪里可以下载 computeGCBias 是吗?
大多数基因组的2bit文件都可以找到 here . 搜索.2bit结尾。否则, Fasta文件可以转换为2bit 使用UCSC程序fatotwobit(可用于不同平台 UCSC here )
deepTools Galaxy <http://deeptools.ie-freiburg.mpg.de> _. |
code @ github <https://github.com/deeptools/deepTools/> _. |