Galaxy相关常见问题
我已经达到了我的配额-我能做些什么来节省一些空间?
确保删除的所有数据集 永久地 从磁盘中删除:转到历史选项按钮,选择“清除已删除的数据集”,然后点击历史面板顶部的“刷新”按钮。
下载完成分析的所有数据集,然后删除这些数据集(单击“x”,然后 确保他们被清除了 (见上文)。
从一个历史记录复制到另一个历史记录对我来说不起作用-数据集根本不显示在目标历史记录中!
- 将数据集从一个历史记录复制到另一个历史记录后,请检查以下两项:
您是否在历史记录面板中看到目的地历史记录,即当前历史记录面板的标题是否与您在主框架中选择的目的地历史记录的名称匹配?
点击刷新按钮
如何使用已发布的工作流?
你 必须注册 如果要使用 deepTools Galaxy . (“用户”-->“注册”-您只需提供一个电子邮件地址)。不过,请务必阅读使用条款!
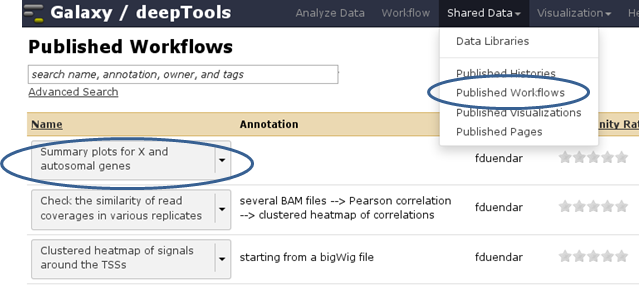
您可以通过“共享数据”-->“已发布的工作流”找到其他用户公开或专门与您共享的工作流。单击您感兴趣的工作流旁边的三角形,然后选择“导入”。
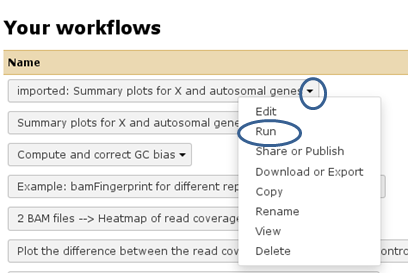
此时会出现一个绿色框,您可以选择“开始使用此工作流”,这将引导您进入自己的工作流菜单(您可以始终通过顶部菜单“工作流”访问)。在这里,您现在应该看到一个标记为“imported:……”的工作流。如果要立即使用工作流,请单击三角形并选择“运行”。工作流现在应该在Galaxy主数据框中可用,并且应该等待您的输入。
我想使用您的工作流程之一-不是在Deeptools星系中,而是在我的研究所提供的本地星系实例中。有可能吗?
是的,这是可能的。唯一的要求是你的本地星系最近安装了一个deeptools。
转到工作流,单击您感兴趣的工作流,然后转到“下载”。这将把工作流保存到您计算机上的.ga文件中。现在转到您的本地Galaxy安装和登录。转到“工作流”菜单并选择“导入工作流”(页面右上角)。单击“浏览”并选择保存的工作流。如果您的本地Galaxy中安装了相同的工具版本,那么这些工作流应该可以立即工作。
plotProfile 说只有当“computematrix运行时——不使用dataaszero”,一个选项才会起作用。我怎样才能知道我是否跑步 computeMatrix 那样?
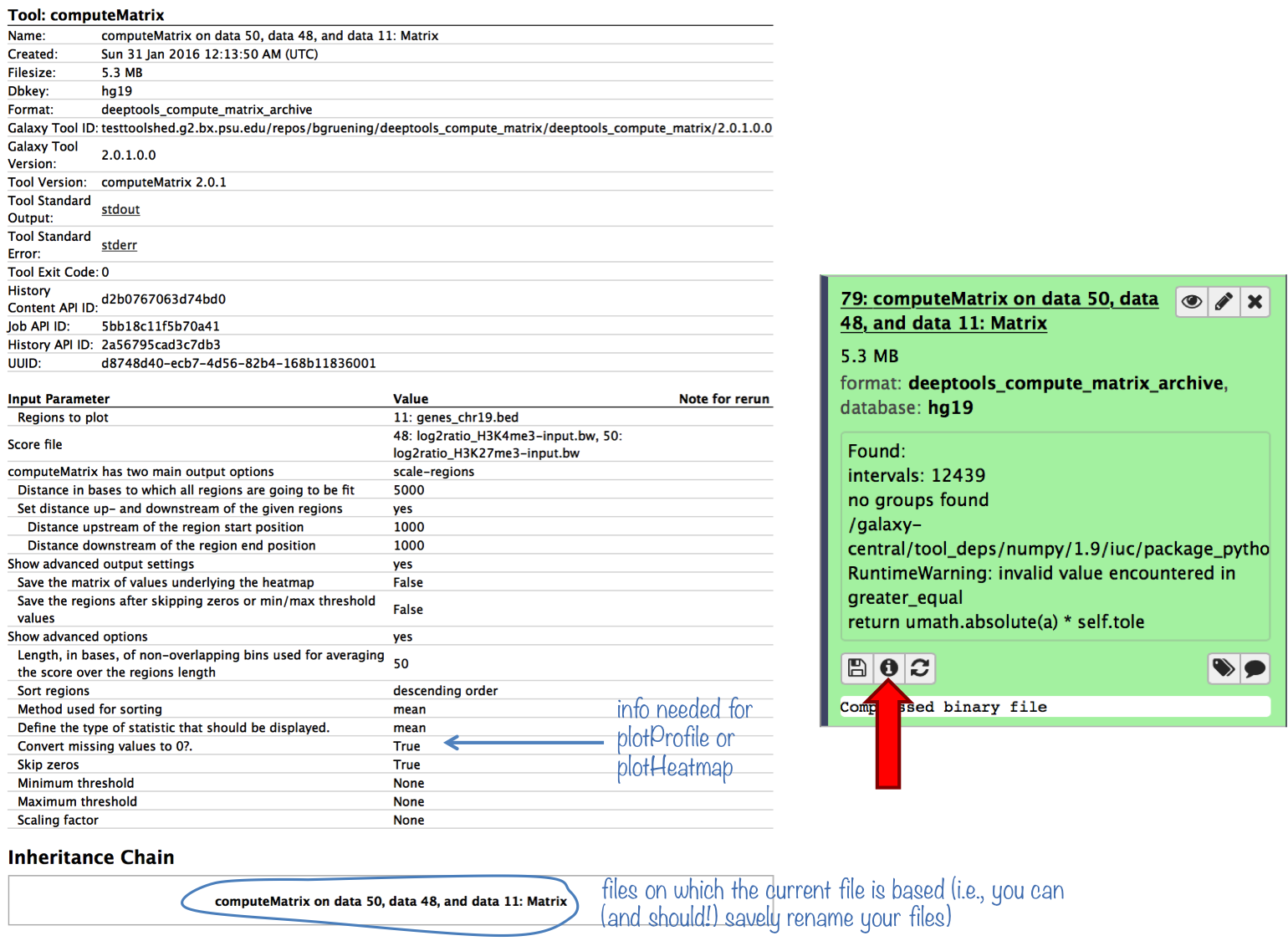
Galaxy会跟踪你所做的一切。要查看您选择生成特定数据集的选项,只需单击“信息”按钮。
我如何才能从Bigwig文件中查看连续读取覆盖率?你推荐哪个基因组浏览器?
有两种流行的基因组浏览器用于可视化连续数据: UCSC 和 IGV .
IGV(推荐)
我们建议下载 IGV ,免费供学术使用。IGV本身需要一个最新的Java安装和相当数量的RAM。它的使用非常直观,而且可以很容易地定制显示。此外,您可以下载全基因组注释数据,这些数据可以与您自己的数据一起显示。
要在IGV中显示数据,请执行以下操作:
访问http://www.broadistitute.org/igv/,注册并下载igv
解包IGV存档并更改为提取的IGV文件夹
使用
igv.bat(Windows)igv.sh(Linux)或igv.command(OSX)启动IGV(有关更多信息,请阅读随附的readme.txt文件或IGV文件)。选择要可视化的文件的基因组版本(如dm3) 这是最重要的一步! IGV不会自动检测基因组版本,即如果您选择mm9,但您的文件是基于人类数据的,它仍然会显示而不显示错误消息(但显然位置错误!)
转到Deeptools Galaxy服务器(http://deeptools.ie-freiburg.mpg.de/)并导航到您选择的数据集
点击你的数据集,你可以看到它的详细信息,如下面的屏幕截图。( 请记住,并非所有数据集都可以在IGV或UCSC中可视化。 我们建议使用 大人物 或 BED 用于可视化的文件。)
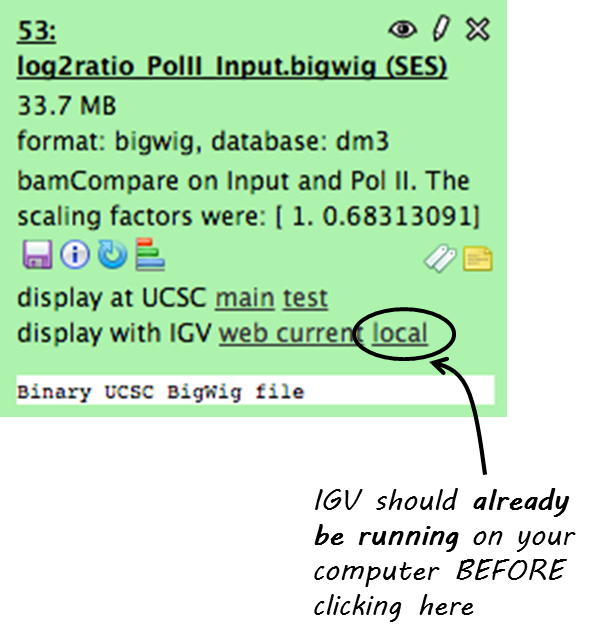
现在单击 “本地显示IGV” 要在IGV中可视化您的数据集,应该已经在您的计算机上运行了。
备注
如果没有安装IGV,则可以使用“带IGV网络电流的显示器”。它将启动一个IGV Web启动版本。 We do *not* recommend that option .
下面是一个典型的bigwig文件显示的屏幕截图:
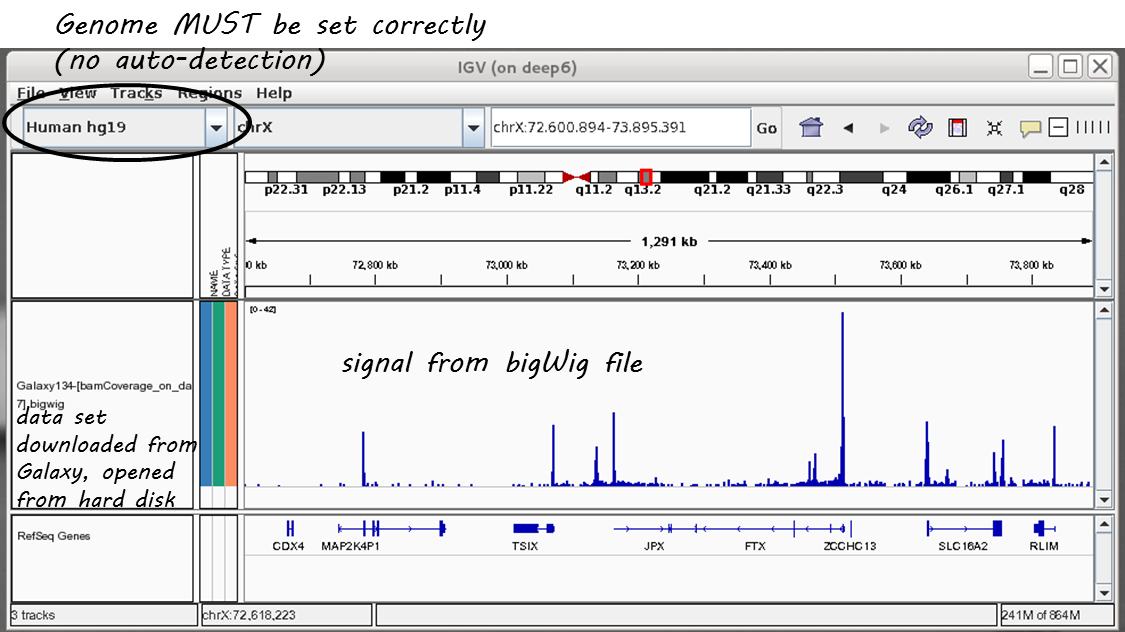
有关详细信息,请查看 IGV documentation .
UCSC
有一个直接的链接从Deeptools星系内部流数据集到UCSC。您可以在数据集图块中找到它:“在UCSC上显示”,如下所示:
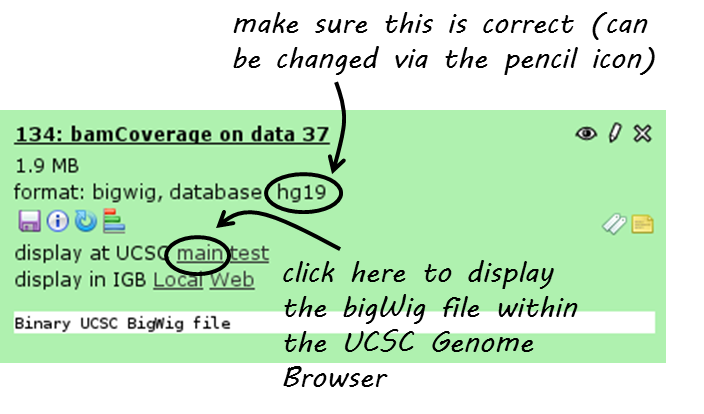
单击“主”,UCSC浏览器将在新窗口中打开,显示您选择的数据集。bigwig文件的默认设置是“密集”显示,看起来像一个热图。
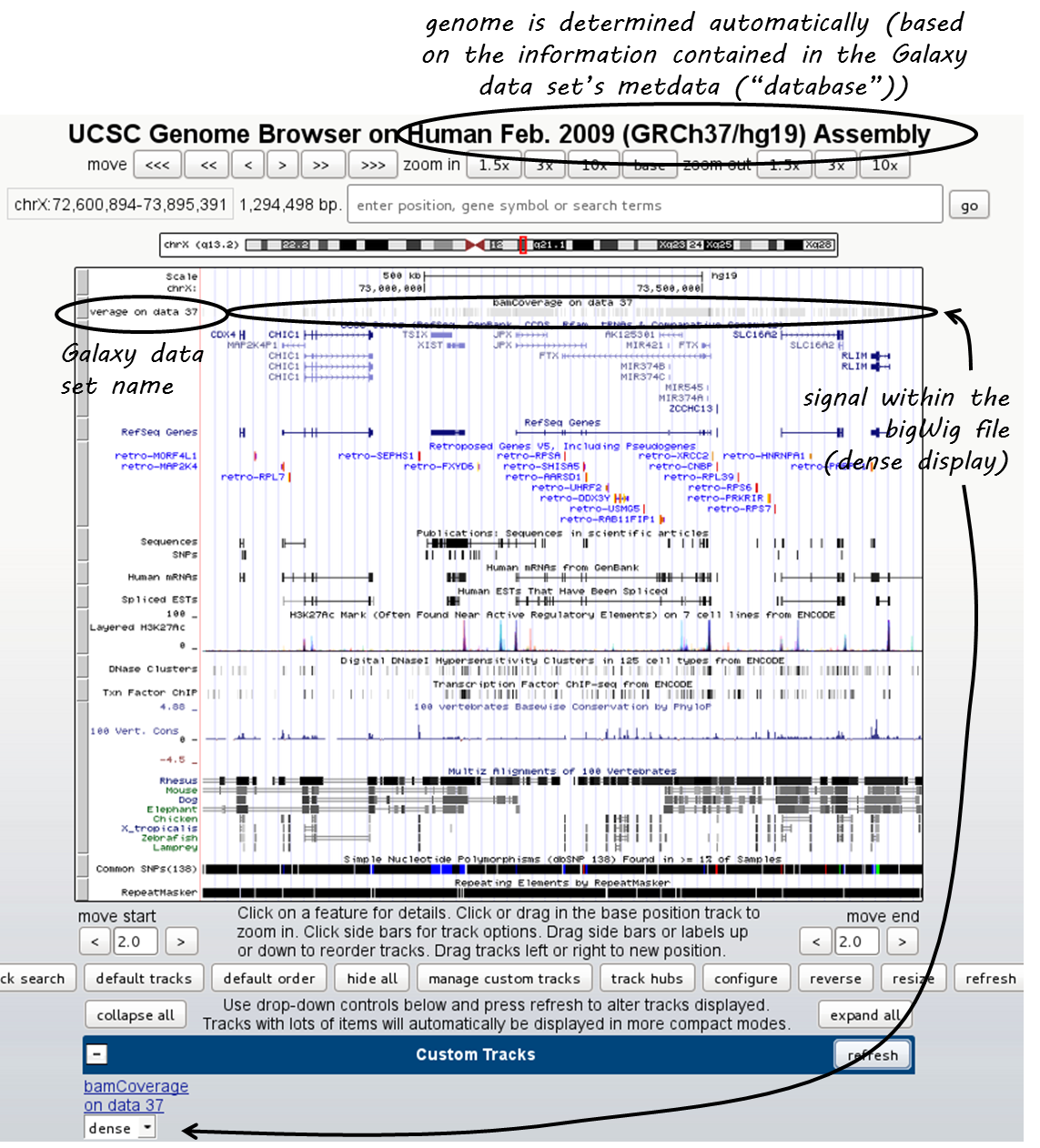
如果您想以类似于IGV屏幕截图中所示的“山谷山”的方式显示连续剖面,请转到自定义轨迹下的下拉菜单并选择“完整”。
UCSC有大量的公共数据,您可以通过向下滚动页面,在自定义曲目条目之外显示这些数据。有关如何使用UCSC基因组浏览器的更多信息,请访问 here .
UCSC的已知问题
染色体命名 :ucsc要求染色体名称以“chr”数字格式表示,例如chr1。如果你把你的读码映射到一个非UCSC标准基因组,很有可能染色体上只标记了它们的编号。UCSC将无法识别从这些BAM文件生成的bigwig文件,即您将看到数据集名称,但没有信号。
没有从硬盘上载Bigwig文件 :为了最小化计算压力,UCSC依赖于流式bigwig文件(即,不需要一次加载整个文件,浏览器将始终只加载用户正在查看的特定区域的数据)。
什么是将deepTool结果与其他下游分析(星系外)相结合的最佳方法?
你可以 保存所有数据表 在Deeptools生成的每个图像的基础上,即如果要以不同的方式绘制平均配置文件,可以下载相应的数据(勾选“高级输出选项”下的相关选项后),并将其导入r、excel、graphpadprism等。
Galaxy中工具的描述还将包含有关如何保存数据以及预期的格式的详细信息。
如何确定BAM文件的基本参数,如读取次数、读取长度、复制速率和平均DNA片段长度?
如果你下载了 BAM 来自公共存储库的文件,很有可能这些特性实际上在那里被注意到了。
如果不是这样的话,我们建议看一下这个工具 FastQC ,将返回上述所有点(片段大小除外)。碎片大小分布可以用deeptools得到。 Bampe碎片大小 (因为Deeptools 2.0)。
deepTools Galaxy <http://deeptools.ie-freiburg.mpg.de> _. |
code @ github <https://github.com/deeptools/deepTools/> _. |