NGS术语表
与大多数专业领域一样,下一代测序也启发了许多首字母缩略词。我们正在努力追踪那些 缩写 我们大量使用。如果有什么不清楚的地方,一定要在 github
缩写
参考基因组通常用其缩写来表示,例如:
hg19=人类基因组,第19版
MM9= 小肌肉 基因组,第9版
DM3= 黑腹果蝇 ,版本3
CE10= 秀丽隐杆线虫 ,版本10
有关可用参考基因组及其缩写的更全面列表,请参见 UCSC data base .
首字母缩略词 |
完整短语 |
同义词/解释 |
|---|---|---|
<anything>-序列 |
-排序 |
表明一个实验是通过DNA测序完成的。 |
芯片序列 |
染色质免疫沉淀测序 |
检测转录因子结合位点和组蛋白修饰的NGS技术(见条目 输入 更多信息) |
DNase |
脱氧核糖核酸酶I |
dnase I消化用于确定活性(“开放”)染色质区域。 |
HTS |
高通量测序技术 |
下一代测序、大规模并行短读测序、深度测序 |
MNase |
微细胞核酸酶 |
用酶消化法测定核小体的位置。 |
NGS |
下一代测序 |
高通量(DNA)测序、大规模并行短读测序、深度测序 |
RPGC |
读取每个基因组内容 |
标准化读取到1X序列深度,序列深度定义为:(映射读取X片段长度)/有效基因组大小 |
RPKM |
每千基读取/百万读取 |
标准化读取数:rpkm(每bin)=reads per bin/(映射读取(百万)x bin长度(kb)) |
对流行音乐的评论 * -Seq应用程序,请参见 Zentner and Henikoff .
NGS和通用术语
以下是一些人可能不熟悉的术语:
箱子
同义词:窗口,区域
“bin”是较大分组的子集。许多计算是通过首先将基因组划分为小区域(bins)来进行的,计算实际上是在小区域(bins)上进行的。
输入
控制实验通常用于芯片序列实验
尽管Chip Seq依赖抗体来丰富与某一蛋白质结合的DNA片段,但输入样本的处理方式应完全相同,不包括抗体。这就可以解释样本处理带来的偏差和细胞的一般染色质结构。
阅读
同义词:标记
这个术语是指由测序者测序(“读取”)的DNA片段。我们试图区分“读取”和“DNA片段”,因为放入测序器的片段往往在200-1000碱基的范围内,其中只有前50-300碱基是典型的测序。大多数deeptools不仅考虑到这些读取,而且扩展它们以匹配原始DNA片段大小。(原始大小将由您给出,或者,如果您使用成对的端点排序,则将从两个读取伙伴之间的距离计算得出)。
文件格式
从下一代测序数据中获得的数据必须进行多次处理。大多数处理步骤的目的都是只提取特定下游分析所需的信息,而冗余条目常常被丢弃。因此, 特定的数据格式通常与数据处理管道的不同步骤相关联。 .
在这里,我们只想给出文件的非常简短的关键描述,为了获得详细的信息,我们将链接到外部网站。请注意,此处的文件名排序是按字母顺序排列的,而不是根据它们在此处描述的分析管道中的用法进行的:
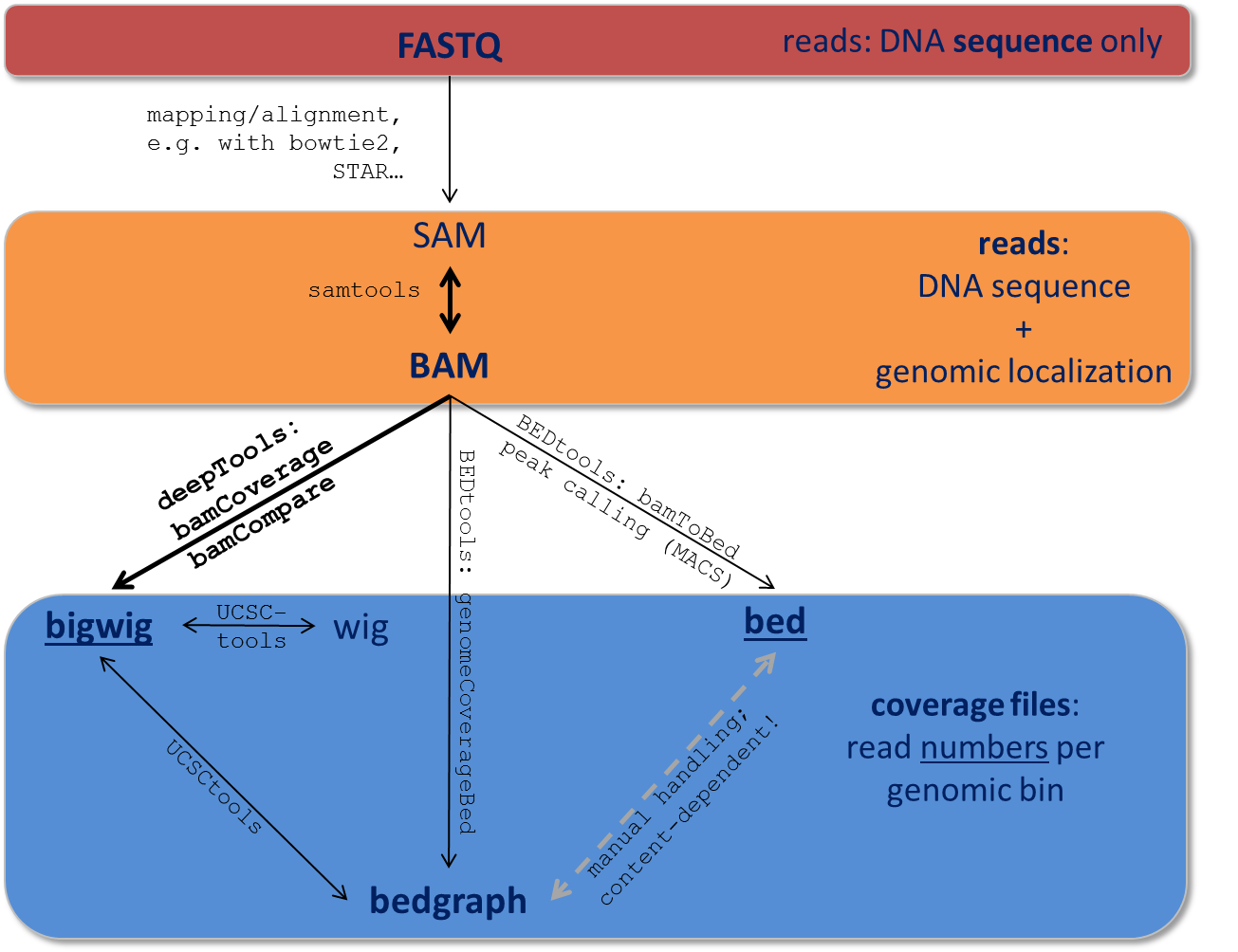
有关图中提到的不同工具集合的更多信息,请遵循链接:
samtools | UCSCtools | BEDtools |
2位
BAM
BED
典型文件扩展名:
.bed文本文件
用于基因组间期,如基因、峰区等。
格式可在 UCSC
对于deeptools,前3列很重要:染色体,区域的起始位置,基因组的结束位置。
不要把它和 床面描记器 格式(尽管它们是相关的)
来自鼠标基因床文件的示例行(请注意,起始位置基于0,结束位置基于1,遵循床文件的UCSC约定)::
chr1 3204562 3661579 NM_001011874 Xkr4 - chr1 4481008 4486494 NM_011441 Sox17 - chr1 4763278 4775807 NM_001177658 Mrpl15 - chr1 4797973 4836816 NM_008866 Lypla1 +
床面描记器
典型文件扩展名:
.bg,.bedGraph文本文件
类似于bed文件(不同!),它可以 only 包含4列和第4列 must 得分
再次阅读 UCSC description 有关详细信息
4个床位图文件的示例行(如遵循UCSC惯例的床位文件,开始位置基于0,结束位置基于1,在床位图文件中):
chr1 10 20 1.5
chr1 20 30 1.7
chr1 30 40 2.0
chr1 40 50 1.8
大人物
典型文件扩展名:
.bw,.bigwig二元的 A的版本 床面描记器 或
wig文件包含间隔和相关分数的坐标
分数可以是任何东西,例如平均阅读覆盖率
UCSC description 有关详细信息
FASTA
典型文件扩展名:
.fasta文本文件,通常为gzip格式 (
.fasta.gz)非常简单的格式 DNA/RNA 或 蛋白质 序列,这可以是从小片段的DNA或蛋白质到整个基因组的任何东西(最有可能的是,你会得到你的有机体对FASTA格式感兴趣的基因组序列)。
见 2位 压缩替代项的文件格式项
示例来自 wikipedia 仅显示一个序列:
>gi|5524211|gb|AAD44166.1| cytochrome b [Elephas maximus maximus]
LCLYTHIGRNIYYGSYLYSETWNTGIMLLLITMATAFMGYVLPWGQMSFWGATVITNLFSAIPYIGTNLV
EWIWGGFSVDKATLNRFFAFHFILPFTMVALAGVHLTFLHETGSNNPLGLTSDSDKIPFHPYYTIKDFLG
LLILILLLLLLALLSPDMLGDPDNHMPADPLNTPLHIKPEWYFLFAYAILRSVPNKLGGVLALFLSIVIL
GLMPFLHTSKHRSMMLRPLSQALFWTLTMDLLTLTWIGSQPVEYPYTIIGQMASILYFSIILAFLPIAGX
IENY
FASTQ
典型文件扩展名:
.fastq,.fq文本文件,通常是gzip(->
.fastq.gz)- 包含原始读取信息--每次读取4行:
读取ID
基本通话
附加信息或空行
排序质量度量-每次基本调用1个
请注意,目前还没有关于读取源于哪里的基因组的信息。
示例来自 wikipedia page ,其中包含更多信息:
@read001 # read ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT # read sequence + # usually empty line !''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65 # ASCII-encoded quality scores
如果您需要了解.fastq文件包含的ASCII编码类型,只需运行 FastQC --它的摘要文件会告诉你
SAM
典型文件扩展名:
.sam通常是序列读取与参考基因组比对的结果。
包含一个短标题部分(条目用@符号标记)和一个对齐部分,其中每行对应一个读(因此,可能有数百万行这样的行)
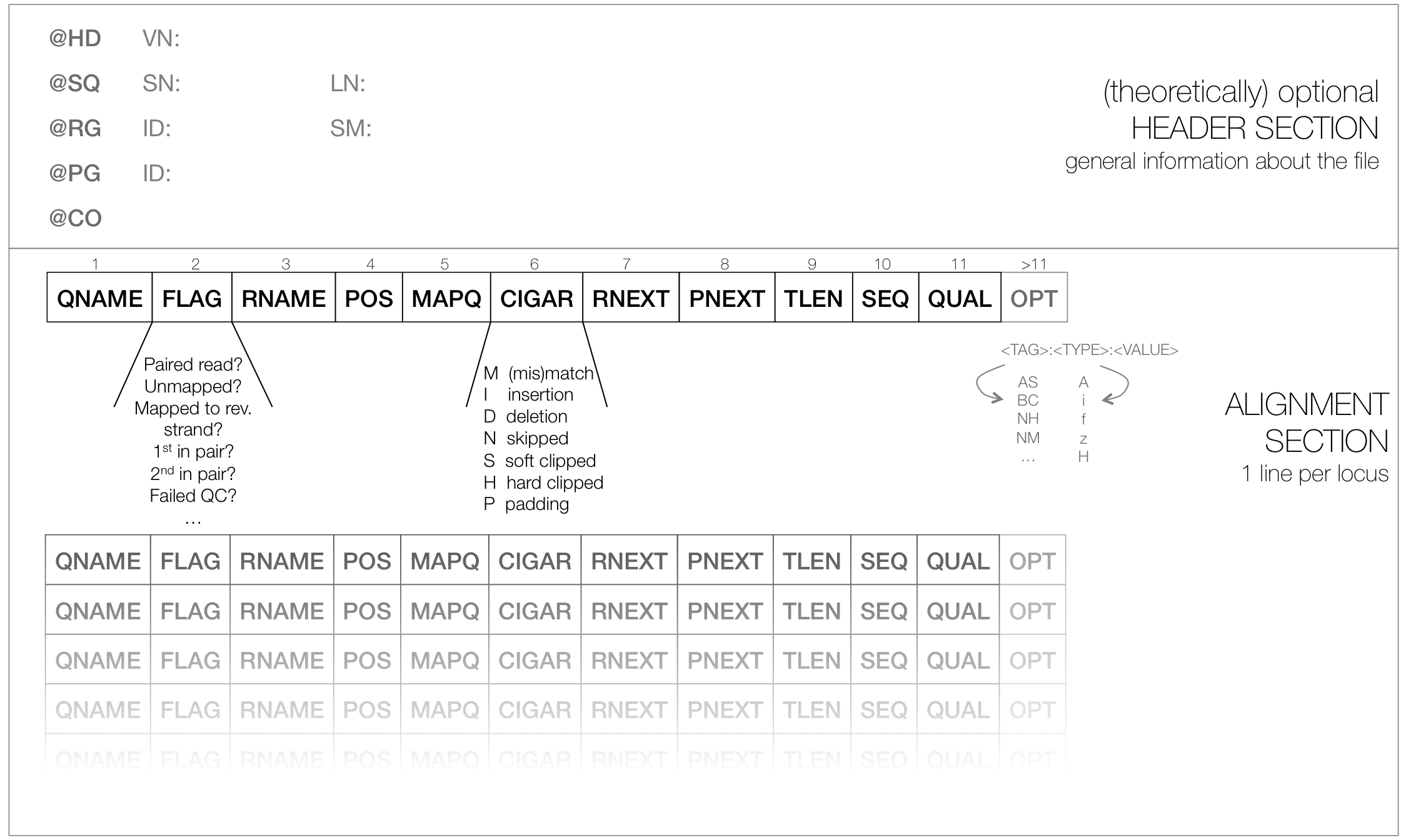
sam头段
制表符分隔的行,以@开头,后跟标记:值对
tag =两个字母的字符串,用于定义 价值
SAM对齐部分
每一行包含有关其绘图质量、序列、在基因组中的位置等信息:
r001 163 chr1 7 30 8M2I4M1D3M = 37 39 TTAGATAAAGGATACTG * r002 0 chr1 9 30 3S6M1P1I4M * 0 0 AAAAGATAAGGATA *这个 第二个字段中的标志 包含对以单个数字编码的几个“是/否”评估的答案
有关标志的更多详细信息,请参阅 this thorough explanation 或 this more technical explanation
这个 第六场雪茄串 表示将读取与特定基因组位置对齐所需的操作类型:
插入
删除(用 D ,更大的删除,例如,对于拼接读取,用表示 N )
剪切(在读取结束时删除)
警告
尽管对SAM/BAM格式进行了相当细致的定义和记录,但是校准程序是否会生成符合这些原则的SAM/BAM文件完全取决于程序员。地图分数,雪茄串,尤其是, 所有可选标志 (字段>11)通常 根据程序的不同定义非常不同 . 如果您计划基于这些条件中的任何一个筛选您的数据,请确保您确切知道这些条目是如何计算和设置的!
deepTools Galaxy <http://deeptools.ie-freiburg.mpg.de> _. |
code @ github <https://github.com/deeptools/deepTools/> _. |