4.7.1.3. 二面角分析 MDAnalysis.analysis.dihedrals
- 作者:
亨利·穆尔
- 年:
2018
- 版权所有:
GNU公共许可证v2
在 0.19.0 版本加入.
此模块包含用于计算给定原子或残基集合的二面角的类。可以对选定的帧或整个轨迹执行此操作。
将生成包含感兴趣角度的时间步长列表,如果需要,可以轻松绘制该列表。对于 Ramachandran 和 Janin 类,可以使用该方法生成基本曲线图 Ramachandran.plot() 或 Janin.plot() 。这些曲线图最好用作参考,但也允许用户自定义。
参见
MDAnalysis.lib.distances.calc_dihedrals()根据原子位置计算二面角的函数
4.7.1.3.1. 示例应用程序
4.7.1.3.1.1. 广义二面体分析
这个 Dihedral 类在计算许多感兴趣的二面体的角度时非常有用。例如,我们可以找到腺苷酸激酶(ADK)5-10残基的phi角。轨迹包含在测试数据文件中:
import MDAnalysis as mda
from MDAnalysisTests.datafiles import GRO, XTC
u = mda.Universe(GRO, XTC)
# selection of atomgroups
ags = [res.phi_selection() for res in u.residues[4:9]]
from MDAnalysis.analysis.dihedrals import Dihedral
R = Dihedral(ags).run()
然后可以通过以下方式访问这些角度 Dihedral.results.angles 。
4.7.1.3.1.2. 拉马钱德兰分析
这个 Ramachandran 类允许快速计算经典的Ramachandran曲线图 [Ramachandran1963] 在脊梁上 \(phi\) 和 \(psi\) 天使。不像 Dihedral 类,该类接受 atomgroups ,这个类只需要残基或这些残基中的原子的列表。前面的示例可以使用以下命令重复:
u = mda.Universe(GRO, XTC)
r = u.select_atoms("resid 5-10")
R = Ramachandran(r).run()
然后可以使用内置的绘图方法进行绘图 Ramachandran.plot() ::
import matplotlib.pyplot as plt
fig, ax = plt.subplots(figsize=plt.figaspect(1))
R.plot(ax=ax, color='k', marker='o', ref=True)
fig.tight_layout()
如本例所示 Ramachandran plot figure 。
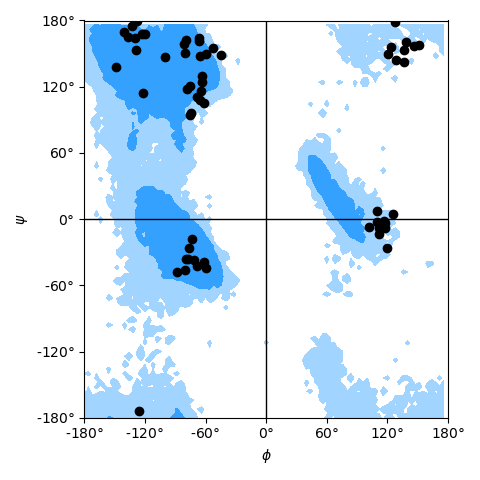
拉马钱德兰绘制了从ADK测试轨迹(XTC)中采样的5至10个ADK残基的拉马钱德兰曲线。背景中的轮廓是“允许的区域”和“边缘允许的”区域。
要自己绘制数据,可以使用以下命令访问角度 Ramachandran.results.angles 。
备注
如果拓扑包含重复或缺失的原子(例如,带有 altloc 或不完全残基)。如果该拓扑具有作为 altloc 属性,则必须仅指定一个 altloc 对于一个以上的原子 ("protein and not altloc B" )。
4.7.1.3.1.3. 贾宁分析
贾宁地块 [Janin1978] 对于侧链构象 (\(\chi_1\) 和 \(chi_2\) 角度)可以使用 Janin 班级。它以同样的方式工作,只需要一个残基列表;请参阅 Janin plot figure 作为一个例子。
角度的数据可以在属性中访问 Janin.results.angles 。
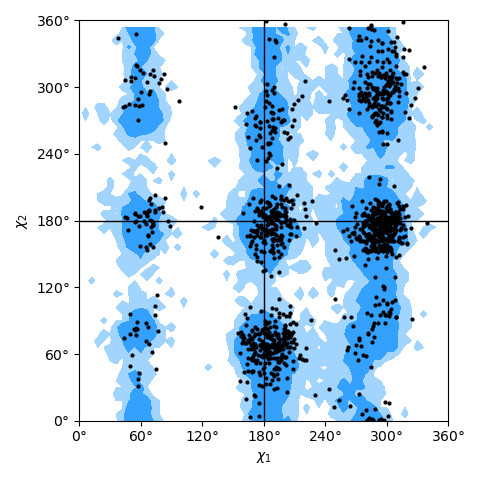
从ADK试验轨迹(XTC)中采样的ADK所有残留物的Janin曲线图。背景中的轮廓是所有可能残留物的“允许区域”和“边缘允许”区域。
备注
如果拓扑包含重复或缺失的原子(例如,带有 altloc 或不完全残基)。如果该拓扑具有作为 altloc 属性,则必须仅指定一个 altloc 对于一个以上的原子 ("protein and not altloc B" )。
此外,许多残留物没有 \(\chi_2\) 如果没有仔细过滤残基的选择,只包括那些带有 both 边链二面体,然后是一个 ValueError 带着这条信息 选择的原子太多或太少 都被养大了。
4.7.1.3.1.4. 参考地块
可以使用Kwarg将参考地块添加到Ramachandran和Janin类的轴线上 ref=True 对于 Ramachandran.plot() 和 Janin.plot() 方法:研究方法。拉马钱德兰参考数据 (Rama_ref )和Janin参考数据 (Janin_ref )使用从500个PDB文件的大量选择中获得的数据来制作,并使用这些类进行分析 [Mull2018] 。Ramachandran参考图的允许和边缘允许区域的分界线设置为包括90%和99%的数据点,Janin参考图的分界线设置为90%和98%的数据点。用于参考图的PDB文件列表取自 [Lovell2003] 和关于一般贾宁地区的信息从 [Janin1978] 。
4.7.1.3.2. 分析类
- class MDAnalysis.analysis.dihedrals.Dihedral(atomgroups, **kwargs)[源代码]
计算指定原子组的二面角。
对于轨迹中的每一步所给出的每个原子群,将计算其二面角。每个
AtomGroup必须包含4个原子。备注
这个类接受列表作为输入,对于大量的原子组选择最有用。如果只有一个感兴趣的原子组,则必须将其作为一个原子组的列表给出。
在 2.0.0 版本发生变更:
angles结果现在存储在MDAnalysis.analysis.base.Results实例。- 参数:
- 抛出:
ValueError -- 如果任何原子基团不包含4个原子
- results.angles
包含列表中每个原子组的角度的时间步长,作为
n_frames×len(atomgroups)numpy.ndarray带内容[[angle 1, angle 2, ...], [time step 2], ...]。在 2.0.0 版本加入.
- angles
的别名
results.angles属性。自 2.0.0 版本弃用: 将在MDAnalysis 3.0.0中删除。请使用
results.angles取而代之的是。
- run(start=None, stop=None, step=None, frames=None, verbose=None, *, progressbar_kwargs={})
执行计算
- 参数:
start (int, optional) -- 分析的起始框架
stop (int, optional) -- 停止分析框架
step (int, optional) -- 要在每个分析的帧之间跳过的帧数
frames (array_like, optional) -- 用于切片轨迹的整数或布尔数组; frames 只能使用 instead 的 start , stop ,以及 step 。设置 both frames 并且至少有一项 start , stop , step 设置为非默认值将引发
ValueError。。。版本已添加::2.2.0verbose (bool, optional) -- 启用详细信息
progressbar_kwargs (dict, optional) -- 带有有关进度条位置等的自定义参数的ProgressBar关键字;请参见
MDAnalysis.lib.log.ProgressBar查看完整的列表。
在 2.2.0 版本发生变更: 添加了分析任意帧的功能,方法是在 frames 关键字参数。
在 2.5.0 版本发生变更: 增列 progressbar_kwargs 参数,允许修改tqdm进度条的描述、位置等
- class MDAnalysis.analysis.dihedrals.Ramachandran(atomgroup, c_name='C', n_name='N', ca_name='CA', check_protein=True, **kwargs)[源代码]
计算 \(\phi\) 和 \(\psi\) 所选残留物的二面角。
\(\phi\) 和 \(\psi\) 将为对应于以下各项的每个残留物计算角度 atomgroup 对于轨迹中的每一个时间步。一个
ResidueGroup从以下位置生成 atomgroup 它与蛋白质进行比较,以确定它是否是合法的选择。- 参数:
示例
对于标准蛋白质,默认参数足以运行Ramachandran分析:
r = Ramachandran(u.select_atoms('protein')).run()
对于含有非标准残基的蛋白质,或用于计算其他线性聚合物的二面角,您可以关闭蛋白质检查,并提供您自己的原子名称来代替典型的肽主干原子:
r = Ramachandran(u.atoms, c_name='CX', n_name='NT', ca_name='S', check_protein=False).run()
上述分析将从Cx‘-Nt-S-Cx的“Phi”选择和Nt-S-Cx-Nt’的“Psi”选择计算角度。
- 抛出:
ValueError -- 如果残基的选择不包含在蛋白质中,并且
check_protein是True
备注
如果
check_protein是True并且残基的选择超出了蛋白质的范围,那么就会出现错误。如果残留物选择包括第一个或最后一个残留物,则会发出警告,并将它们从残留物列表中删除,但分析仍将运行。如果一个 \(\phi\) 或 \(\psi\) 无法进行选择,该残留物将从分析中移除。在 1.0.0 版本发生变更: 添加了c_name、n_name、ca_name和check_Protein关键字参数
在 2.0.0 版本发生变更:
angles结果现在存储在MDAnalysis.analysis.base.Results实例。- results.angles
的时间步长。 \(\phi\) 和 \(\psi\) 每个残留物的角度作为
n_frames×n_residues×2numpy.ndarray带内容[[[phi, psi], [residue 2], ...], [time step 2], ...]。在 2.0.0 版本加入.
- angles
的别名
results.angles属性。自 2.0.0 版本弃用: 将在MDAnalysis 3.0.0中删除。请使用
results.angles取而代之的是。
- plot(ax=None, ref=False, **kwargs)[源代码]
将数据绘制成标准的Ramachandran曲线图。
每一次步入
Ramachandran.results.angles都绘制在同一张图上。- 参数:
ax (
matplotlib.axes.Axes) -- 如果没有 ax 被提供或设置为None然后,该绘图将被添加到当前活动轴。ref (bool, optional) -- 添加显示允许区域和边缘允许区域的常规Ramachandran图
kwargs (optional) -- 所有其他的科瓦格都被传给了
matplotlib.pyplot.scatter()。
- 返回:
ax --带有情节的轴线 ax 或者是当前的轴线。
- 返回类型:
- run(start=None, stop=None, step=None, frames=None, verbose=None, *, progressbar_kwargs={})
执行计算
- 参数:
start (int, optional) -- 分析的起始框架
stop (int, optional) -- 停止分析框架
step (int, optional) -- 要在每个分析的帧之间跳过的帧数
frames (array_like, optional) -- 用于切片轨迹的整数或布尔数组; frames 只能使用 instead 的 start , stop ,以及 step 。设置 both frames 并且至少有一项 start , stop , step 设置为非默认值将引发
ValueError。。。版本已添加::2.2.0verbose (bool, optional) -- 启用详细信息
progressbar_kwargs (dict, optional) -- 带有有关进度条位置等的自定义参数的ProgressBar关键字;请参见
MDAnalysis.lib.log.ProgressBar查看完整的列表。
在 2.2.0 版本发生变更: 添加了分析任意帧的功能,方法是在 frames 关键字参数。
在 2.5.0 版本发生变更: 增列 progressbar_kwargs 参数,允许修改tqdm进度条的描述、位置等
- class MDAnalysis.analysis.dihedrals.Janin(atomgroup, select_remove='resname ALA CYS* GLY PRO SER THR VAL', select_protein='protein', **kwargs)[源代码]
计算 \(\chi_1\) 和 \(\chi_2\) 所选残留物的二面角。
\(\chi_1\) 和 \(\chi_2\) 将为对应于以下各项的每个残留物计算角度 atomgroup 对于轨迹中的每一个时间步。一个
ResidueGroup从以下位置生成 atomgroup 它与蛋白质进行比较,以确定它是否是合法的选择。备注
如果残基选择超出了蛋白质的范围,则会出现错误。如果残基选择包括残基Ala、Cys*、Gly、PRO、SER、Thr或Val(默认为 select_remove 关键字参数),则将发出警告,并将它们从残留物列表中移除,但分析仍将运行。某些拓扑具有altloc属性,这些属性可能会向选择中添加重复的原子,因此必须将其删除。
- 参数:
atomgroup (AtomGroup or ResidueGroup) -- 用于残基的原子 \(\chi_1\) 和 \(\chi_2\) 是经过计算的
select_remove (str) -- 选择字符串以移除不具有 \(chi_2\) 角度
select_protein (str) -- 从中再选择仅含蛋白质残基的选择字符串 atomgroup 检查是否只选择了氨基酸;如果您有非标准氨基酸,则调整此选择以包括它们
- 抛出:
ValueError -- 如果最终选择的残基不包含在蛋白质中(由
atomgroup.select_atoms(select_protein))ValueError -- 如果在选择中为残基找到的原子不够多或太多,通常是由于缺少原子或替代位置,或由于非标准残基
在 2.0.0 版本发生变更: select_remove 和 select_protein 添加了关键字。
angles结果现在存储在MDAnalysis.analysis.base.Results实例。- results.angles
的时间步长。 \(\chi_1\) 和 \(\chi_2\) 每个残留物的角度作为
n_frames×n_residues×2numpy.ndarray带内容[[[chi1, chi2], [residue 2], ...], [time step 2], ...]。在 2.0.0 版本加入.
- angles
的别名
results.angles属性。自 2.0.0 版本弃用: 将在MDAnalysis 3.0.0中删除。请使用
results.angles取而代之的是。
- plot(ax=None, ref=False, **kwargs)[源代码]
将数据打印到标准的Janin图中。
每一次步入
Janin.results.angles都绘制在同一张图上。- 参数:
ax (
matplotlib.axes.Axes) -- 如果没有 ax 被提供或设置为None然后,该绘图将被添加到当前活动轴。ref (bool, optional) -- 添加显示允许区域和边缘允许区域的一般Janin图
kwargs (optional) -- 所有其他的科瓦格都被传给了
matplotlib.pyplot.scatter()。
- 返回:
ax --带有情节的轴线 ax 或者是当前的轴线。
- 返回类型:
- run(start=None, stop=None, step=None, frames=None, verbose=None, *, progressbar_kwargs={})
执行计算
- 参数:
start (int, optional) -- 分析的起始框架
stop (int, optional) -- 停止分析框架
step (int, optional) -- 要在每个分析的帧之间跳过的帧数
frames (array_like, optional) -- 用于切片轨迹的整数或布尔数组; frames 只能使用 instead 的 start , stop ,以及 step 。设置 both frames 并且至少有一项 start , stop , step 设置为非默认值将引发
ValueError。。。版本已添加::2.2.0verbose (bool, optional) -- 启用详细信息
progressbar_kwargs (dict, optional) -- 带有有关进度条位置等的自定义参数的ProgressBar关键字;请参见
MDAnalysis.lib.log.ProgressBar查看完整的列表。
在 2.2.0 版本发生变更: 添加了分析任意帧的功能,方法是在 frames 关键字参数。
在 2.5.0 版本发生变更: 增列 progressbar_kwargs 参数,允许修改tqdm进度条的描述、位置等
引用
G.N. Ramachandran, C. Ramakrishnan, and V. Sasisekharan. Stereochemistry of polypeptide chain configurations. Journal of Molecular Biology, 7(1):95–99, 1963. URL: https://www.sciencedirect.com/science/article/pii/S0022283663800236, doi:https://doi.org/10.1016/S0022-2836(63)80023-6.
Joël Janin, Shoshanna Wodak, Michael Levitt, and Bernard Maigret. Conformation of amino acid side-chains in proteins. Journal of Molecular Biology, 125(3):357–386, 1978. URL: https://www.sciencedirect.com/science/article/pii/0022283678904084, doi:https://doi.org/10.1016/0022-2836(78)90408-4.
Henry Mull and Oliver Beckstein. Technical Report: SPIDAL Summer REU 2018 Dihedral Analysis in MDAnalysis. 8 2018. URL: https://figshare.com/articles/journal_contribution/Technical_Report_SPIDAL_Summer_REU_2018_Dihedral_Analysis_in_MDAnalysis/6957296, doi:10.6084/m9.figshare.6957296.v1.
Simon C. Lovell, Ian W. Davis, W. Bryan Arendall III, Paul I. W. de Bakker, J. Michael Word, Michael G. Prisant, Jane S. Richardson, and David C. Richardson. Structure validation by c$/alpha$ geometry: ψ,φ and cβ deviation. Proteins: Structure, Function, and Bioinformatics, 50(3):437–450, 2003. doi:https://doi.org/10.1002/prot.10286.