复基因分析
默认情况下, computeMatrix 在整个相邻区域(如转录本)上使用信号计算其输出。虽然这通常非常有用,但在诸如rnaseq的情况下,结果并不理想。以下面的基因模型和覆盖率概况为例:
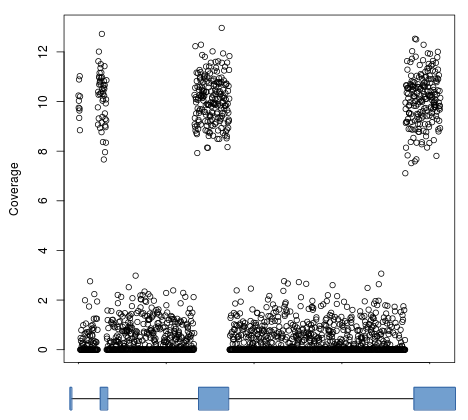
如果使用这种块状覆盖进行聚类,那么结果将受到外显子数量及其位置的影响。相反,人们通常希望忽略内含子区域,只使用外显子中的信号(由基因模型中的区块表示)。这可以通过使用 --metagene 选择权 computeMatrix 并提供BED12或GTF文件作为一组区域:
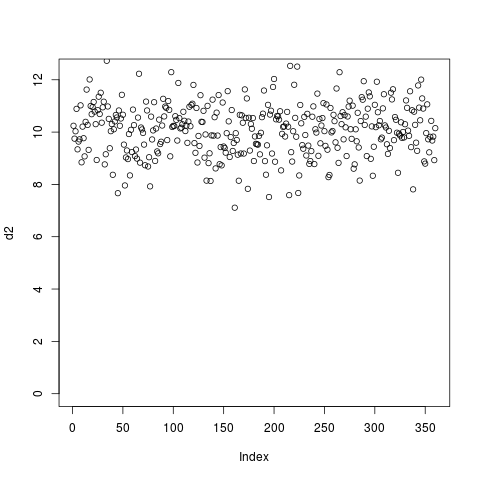
注意,对于GTF文件,用于定义外显子的区域可以很容易地修改。例如,对于riboseq样本,最好使用带注释的编码区域,因此指定 --exonID CDS . 同样,整个基因可以通过指定 --transcriptID gene --transcript_id_designator gene_id .
deepTools Galaxy <http://deeptools.ie-freiburg.mpg.de> _. |
code @ github <https://github.com/deeptools/deepTools/> _. |