黑名单地区
Chipseq实验中存在许多偏差源。其中最普遍的是来自“黑名单”区域的信号(参见 Carroll et al. 以及其中的历史背景参考)。黑名单区域在许多芯片实验类型中显示出显著的丰富信号(例如,无论是什么IPED或实验条件)。包括这些区域不仅会导致假阳性峰值,而且还会在样本归一化之间产生偏差。示例如下:
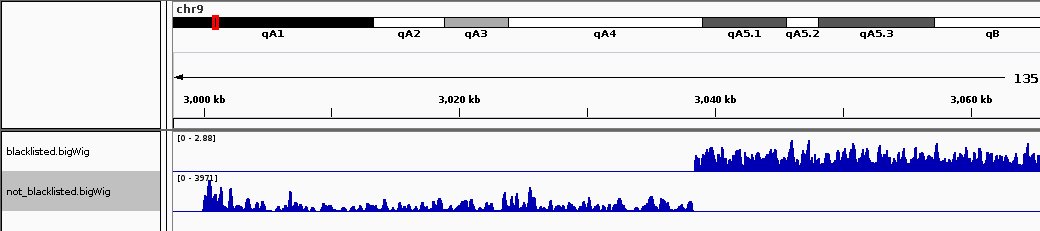
9号染色体上从300万左右开始的区域标志着一个带注释的卫星重复序列的开始。由于该区域包含的读数比预期的要大得多,因此样本之间的富集程度的细微差异可能会导致样本缩放之间的错误,从而掩盖非重复区域中的信号。这可以在下面的IGV屏幕截图中看到,黑名单区域就在屏幕的一侧。
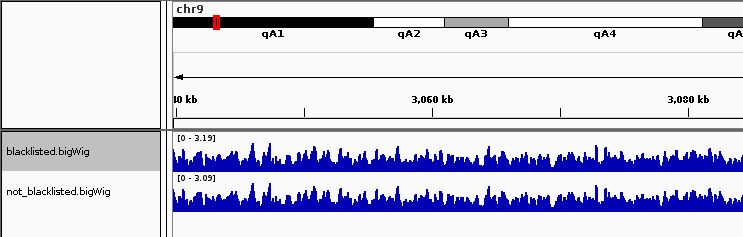
请注意,由于黑名单区域的存在,黑名单区域之外的信号会稍微降低。使用 --blackListFileName 整个Deeptools都提供选项。在所有标准化中,这些区域的减法都被考虑在内。
备注
一些程序,例如 bamCoverage ,可以使用文件中的读取次数来估计覆盖率,以便进行规范化。为此,将减去黑名单区域中的读取次数,但请注意,黑名单区域中的读取次数是通过计算每个区域中完全包含的读取次数来计算的。因此,如果使用重叠的区域,则可能会对将被列入黑名单的读取数进行双重计数,从而降低规范化的可靠性。从2.5.5版开始,如果黑名单区域重叠,deeptools将打印警告并退出。如果其中任何一个接近1千字节,它也会发出警告,因为这在生物学上不太可能是合理的(实际上,它可能低估了实际应该列入黑名单的读取次数和区域)。
deepTools Galaxy <http://deeptools.ie-freiburg.mpg.de> _. |
code @ github <https://github.com/deeptools/deepTools/> _. |