Deeptools API示例
下面简要概述了deeptools中最有用的方法和类。完整信息可在以下链接中找到: 索引 和 模块索引
在区域中查找读取覆盖范围
使用deeptools,可以使用多个处理器快速计算多个基因组区域和多个文件的读取覆盖率。首先,我们从一个简单的例子开始,随后对其进行了扩展,以演示多处理器的使用。在这个例子中,我们计算了50bp的容器在一个小区域上的读取覆盖率。为此,我们需要 deeptools.countReadsPerBin 班级。
import deeptools.countReadsPerBin as crpb
我们还需要一个包含对齐读取的BAM文件。必须为BAM文件编制索引,以允许快速访问落入感兴趣区域的读取。
bam_file = "file.bam"
现在, CountReadsPerBin 无法初始化对象。构造函数的第一个参数是一个BAM文件列表,在本例中,它只是一个文件。我们将使用 binLength 在50个基地中,随后的垃圾箱相邻(即 stepSize 箱子之间也有50个底座)。可通过设置 stepSize 小于 binLength .
cr = crpb.CountReadsPerBin([bam_file], binLength=50, stepSize=50)
现在,我们可以计算从0到1000的2号染色体区域的覆盖率。
cr.count_reads_in_region('chr2L', 0, 1000)
(array([[ 2.],
[ 3.],
[ 1.],
[ 2.],
[ 3.],
[ 2.],
[ 4.],
[ 3.],
[ 2.],
[ 3.],
[ 4.],
[ 6.],
[ 4.],
[ 2.],
[ 2.],
[ 1.]]), '')
结果是一个元组,第一个元素是一个numpy数组,每个bin一行,每个BAM文件一列。因为只使用了一个BAM文件,所以只有一列。如果指定了用于保存原始数据的文件名,则用于此操作的临时文件名将显示在元组的第二项中。
过滤读取
如果应该筛选读取,则只需将相关选项传递给构造函数。在以下代码中,对读取进行过滤,以便仅保留映射质量至少为20且未与反向链对齐的那些读取(samflag_exclude=16,其中16是反向读取的值,请参见 [山姆旗计算器] (http://broadistitute.github.io/picard/explain-flags.html)了解更多信息)。此外,重复读取被忽略。
cr = crpb.CountReadsPerBin([bam_file], binLength=50, stepSize=50,
minMappingQuality=20,
samFlag_exclude=16,
ignoreDuplicates=True
)
cr.count_reads_in_region('chr2L', 1000000, 1001000)
(array([[ 1.],
[ 1.],
[ 0.],
[ 0.],
[ 0.],
[ 0.],
[ 2.],
[ 3.],
[ 1.],
[ 0.],
[ 1.],
[ 2.],
[ 0.],
[ 0.],
[ 1.],
[ 2.],
[ 1.],
[ 0.],
[ 0.],
[ 0.]]), '')
基因组取样
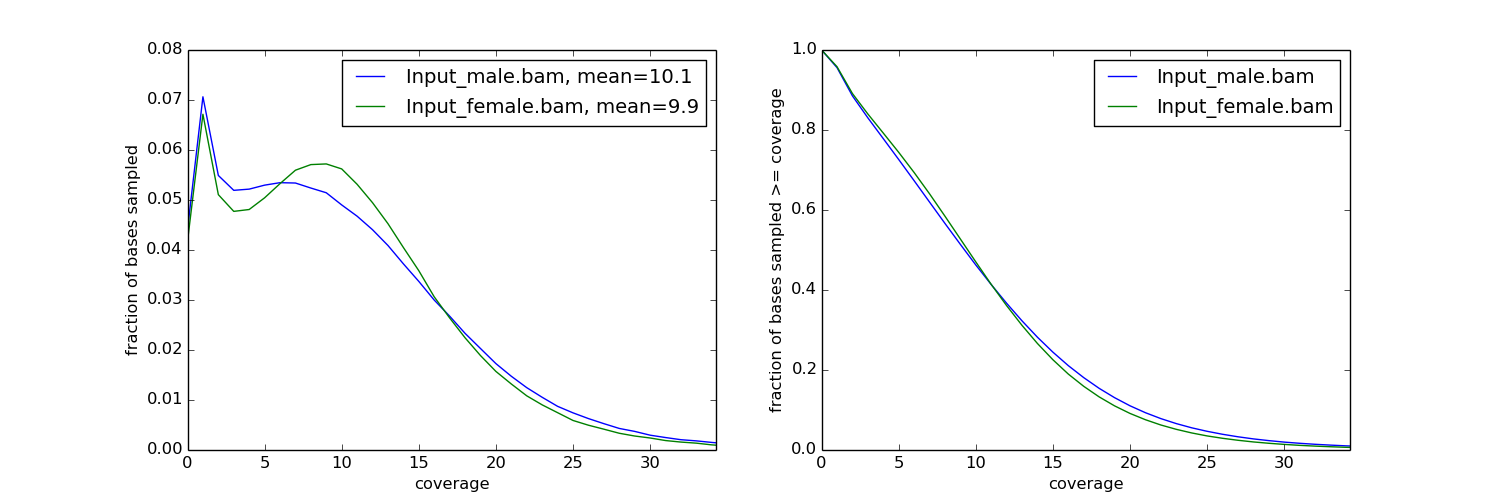
与以前的情况不同,基因组可以简单地取样,而不是相邻的箱。这有助于估计一些值,如测序深度,而无需查看完整的基因组。在下面的示例中,将从三个BAM文件中查询10000个大小为1的基位置,以计算平均排序深度。为此,我们设置了 numberOfSamples 对象构造函数中的参数。
这个 run() 方法而不是 count_reads_in_region 提供整个基因组的有效取样。
cr = crpb.CountReadsPerBin([bam_file1, bam_file2, bam_file3],
binLength=1, numberOfSamples=10000,
numberOfProcessors=10)
sequencing_depth = cr.run()
print sequencing_depth.mean(axis=0)
[ 1.98923924 2.43743744 22.90102603]
这个 run() 方法将计算拆分为10个处理器,并对结果进行比较。当参数 numberOfSamples 使用时,为计算覆盖范围选择的区域不是随机的。相反,基因组被分割成“样本数量”相等的部分,并查询每个部分的起始部分以获取其覆盖范围。您还可以通过输入床位文件来计算所选区域的覆盖率。
现在可以根据结果绘制一些诊断图:
fig, axs = plt.subplots(1, 2, figsize=(15,5))
# plot coverage
for col in res.T:
axs[0].plot(np.bincount(col.astype(int)).astype(float)/total_sites)
csum = np.bincount(col.astype(int))[::-1].cumsum()
axs[1].plot(csum.astype(float)[::-1] / csum.max())
axs[0].set_xlabel('coverage')
axs[0].set_ylabel('fraction of bases sampled')
# plot cumulative coverage
axs[1].set_xlabel('coverage')
axs[1].set_ylabel('fraction of bases sampled >= coverage')
计算frip分数
frip分数被定义为读到峰值的部分,通常用作chip seq质量的度量。对于这个例子,我们需要一个包含峰值区域的bed文件。这些文件通常使用峰值调用者来计算。另外,将使用两个BAM文件,对应于两个生物复制。
bed_files = ["peaks.bed"]
cr = countReadsPerBin.CountReadsPerBin([bam_file1, bam_file2],
bedFile=bed_files,
numberOfProcessors=10)
reads_at_peaks = cr.run()
print reads_at_peaks
array([[ 322., 248.],
[ 231., 182.],
[ 112., 422.],
...,
[ 120., 76.],
[ 235., 341.],
[ 246., 265.]])
结果是一个numpy数组,每个峰值区域有一行,每个BAM文件有一列。
reads_at_peaks.shape
(6295, 2)
现在,计算每个BAM文件每个峰值的读取总数:
total = reads_at_peaks.sum(axis=0)
接下来,我们需要找到每个BAM文件中映射读取的总数。为此,我们使用pysam模块。
import pysam
bam1 = pysam.AlignmentFile(bam_file1)
bam2 = pysam.AlignmentFile(bam_file2)
现在, bam1.mapped 和 bam2.mapped 分别包含每个BAM文件中映射读取的总数。
最后,我们可以计算FRIP分数:
frip1 = float(total[0]) / bam1.mapped
frip2 = float(total[1]) / bam2.mapped
print frip1, frip2
0.170030741997, 0.216740390353
使用mapreduce对成对的末端片段长度进行采样
deeptools在内部使用map reduce策略,在这种策略中,计算被分割成更小的部分,然后发送到不同的处理器。随后对来自不同处理器的输出进行整理。下面的示例基于 bamPEFragmentSize.py
在这里,我们从一个BAM文件中检索读取数据并收集片段长度。读取是使用pysam检索的,并且 read 返回的对象包含 template_length 属性,它是读取对中从最左边到最右边的映射基的数目。
首先,我们将创建一个函数,它可以从一个BAM文件收集基因组位置上的片段长度。正如我们稍后将使用mapreduce调用此函数一样,该函数只接受一个参数,即带有参数的元组:染色体名称、起始位置、结束位置和BAM文件名。
import pysam
import numpy as np
def get_fragment_length(args):
chrom, start, end, bam_file_name = args
bam = pysam.AlignmentFile(bam_file_name)
f_lens_list = []
for fetch_start in range(start, end, 1000000):
# simply get the reads over a region of 10000 bases
fetch_end = min(end, fetch_start + 10000)
f_lens_list.append(np.array([abs(read.template_length)
for read in bam.fetch(chrom, fetch_start, fetch_end)
if read.is_proper_pair and read.is_read1]))
# concatenate all results
return np.concatenate(f_lens_list)
现在,我们可以使用 mapReduce 调用这个函数并计算整个基因组的片段长度。MapReduce需要知道染色体大小,这可以很容易地从BAM文件中检索到。此外,它需要知道发送到每个处理器的区域的大小。在本例中,使用 genomeChunkLength 参数。换句话说,每个处理器执行相同的 get_fragment_length 函数用于收集不同1000万个基本区域的数据。mapreduce的参数是除了第一个必需的三个参数(chrom start,end)之外,发送给函数的参数列表。在这种情况下,只有一个额外的参数传递给函数,即BAM文件名。接下来的两个位置参数是要调用的函数的名称 (get_fragment_length )以及染色体的大小。
import deeptools.mapReduce
bam = pysam.AlignmentFile(bamFile)
chroms_sizes = list(zip(bam.references, bam.lengths))
result = mapReduce.mapReduce([bam_file_name],
get_fragment_length,
chroms_sizes,
genomeChunkLength=10000000,
numberOfProcessors=20,
verbose=True)
fragment_lengths = np.concatenate(result)
print("mean fragment length {}".format(fragment_lengths.mean()))
print("median fragment length {}".format(np.median(fragment_lengths)))
0.170030741997, 0.216740390353
索引和表格
deepTools Galaxy <http://deeptools.ie-freiburg.mpg.de> _. |
code @ github <https://github.com/deeptools/deepTools/> _. |