系统发生学与Bio.Phylo
Biopython 1.54中引入了Bio.Phylo模块。它遵循SeqIO和AlignIO的领导,旨在提供一种独立于源数据格式的系统发生树的通用方法,以及用于I/O操作的一致API。
Bio.Phylo在一篇开放获取期刊文章Talevich中进行了描述 et al. 2012 [Talevich2012], 您可能也会发现这很有帮助。
演示:树里有什么?
为了熟悉该模块,让我们从已经构建的树开始,并用几种不同的方法检查它。然后我们将对分支进行着色,以使用特殊的MIDI HTML功能,并最后保存它。
创建一个名为 simple.dnd using your favorite text editor, or use simple.dnd Biopython源代码提供:
(((A,B),(C,D)),(E,F,G));
该树没有分支长度,只有一个布局和标记的终端。(If您有一个真正的树文件可用,您可以使用它来观看此演示。)
启动您选择的Python解释器:
$ ipython -pylab
对于交互式工作,使用 -pylab 标志启用 matplotlib 集成,因此图形会自动弹出。我们将在此演示中使用它。
现在,在Python中,读取树文件,给出文件名和格式名称。
>>> from Bio import Phylo
>>> tree = Phylo.read("simple.dnd", "newick")
将树对象打印为字符串可以让我们看到整个对象层次结构。
>>> print(tree)
Tree(rooted=False, weight=1.0)
Clade()
Clade()
Clade()
Clade(name='A')
Clade(name='B')
Clade()
Clade(name='C')
Clade(name='D')
Clade()
Clade(name='E')
Clade(name='F')
Clade(name='G')
的 Tree 对象包含有关树的全局信息,例如它是有根的还是无根的。它有一个根分支,在该分支之下,是一直到尖端的嵌套分支列表。
功能 draw_ascii 创建一个简单的ASCII艺术(纯文本)树图。这是交互式探索的方便可视化,以防没有更好的图形工具可用。
>>> from Bio import Phylo
>>> tree = Phylo.read("simple.dnd", "newick")
>>> Phylo.draw_ascii(tree)
________________________ A
________________________|
| |________________________ B
________________________|
| | ________________________ C
| |________________________|
_| |________________________ D
|
| ________________________ E
| |
|________________________|________________________ F
|
|________________________ G
如果你有 matplotlib 或 pylab 安装后,您可以使用创建图形树 draw 功能
>>> tree.rooted = True
>>> Phylo.draw(tree)
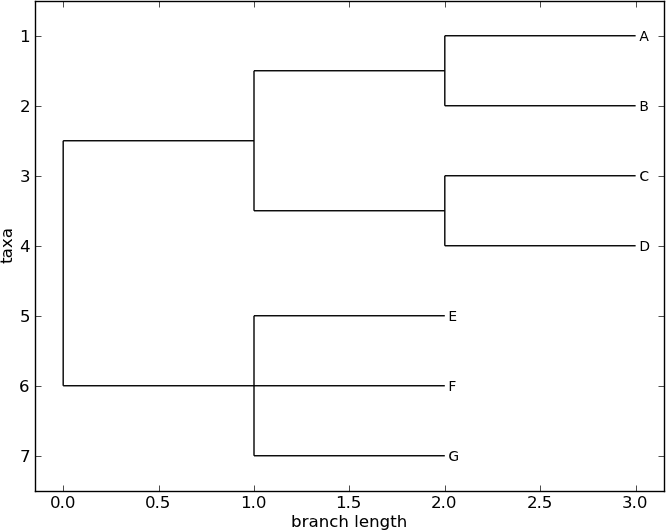
图 6 一棵扎根的树,画着 Phylo.draw .
看到 图 6 .
为树上的树枝上色
功能 draw 支持树中不同颜色和树枝宽度的显示。截至Biopython 1.59, color 和 width 属性在基本Clade对象上可用,并且无需额外使用它们。这两个属性都引用给定分支的领导分支,并以递进方式应用,因此所有后代分支也将在显示期间继承指定的宽度和颜色值。
在Biopython的早期版本中,这些都是PhyloML树的特殊功能,使用属性需要首先将树转换为Bio.Phylo. PhyloML模块中名为Phylogy的基本树对象的子集。
在Biopython 1.55及更高版本中,这是一种方便的树方法:
>>> tree = tree.as_phyloxml()
在Biopython 1.54中,您可以通过一个额外的导入来完成同样的事情:
>>> from Bio.Phylo.PhyloXML import Phylogeny
>>> tree = Phylogeny.from_tree(tree)
请注意,文件格式Newick和Nexus不支持分支颜色或宽度,因此如果您在Bio.Phylo中使用这些属性,则您将只能以PhyloML格式保存值。(You仍然可以将树保存为Newick或Nexus,但输出文件中将跳过颜色和宽度值。)
现在我们可以开始分配颜色了。首先,我们将根进化枝染成灰色。我们可以通过将24位颜色值指定为一个Ruby三重组、一个HTML风格的十六进制字符串或一个预定义颜色的名称来做到这一点。
>>> tree.root.color = (128, 128, 128)
或者:
>>> tree.root.color = "#808080"
或者:
>>> tree.root.color = "gray"
进化枝的颜色被视为通过整个进化枝向下层叠,因此当我们在这里给根上色时,整棵树就会变成灰色。我们可以通过在树的下方指定不同的颜色来覆盖它。
让我们瞄准名为“E”和“F”的节点的最近共同祖先(MRCA)。的 common_ancestor 方法返回原始树中该进化枝的引用,因此当我们为该进化枝“salmon”着色时,该颜色将显示在原始树中。
>>> mrca = tree.common_ancestor({"name": "E"}, {"name": "F"})
>>> mrca.color = "salmon"
如果我们恰好确切地知道某个分支在树中的位置,就嵌套列表条目而言,我们可以通过索引它直接跳转到树中的该位置。这里,索引 [0,1] 指的是根的第一个孩子的第二个孩子。
>>> tree.clade[0, 1].color = "blue"
最后,展示我们的作品:
>>> Phylo.draw(tree)
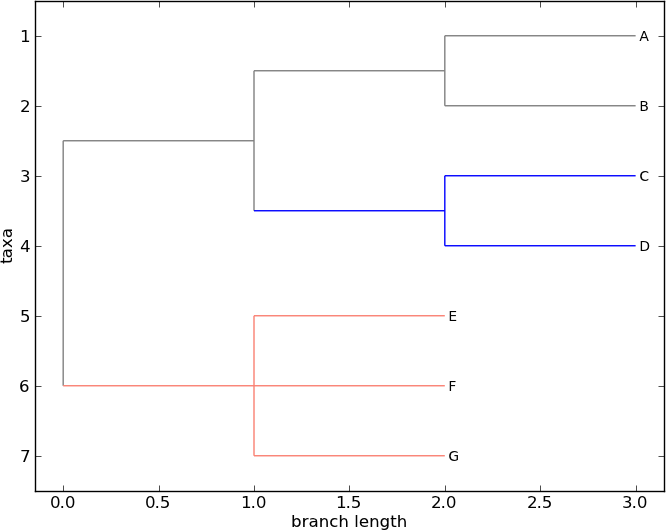
图 7 一棵彩色树,用 Phylo.draw .
看到 图 7 .
请注意,进化枝的颜色包括通向该进化枝的分支及其后代。E和F的共同祖先原来就在根部下方,通过这种颜色我们可以准确地看到树根的位置。
天啊,我们完成了很多!我们休息一下,把工作留着。叫 write 具有文件名或手柄的函数-在这里我们使用标准输出,看看会写什么-以及格式 phyloxml . PhyloML保存了我们分配的颜色,因此您可以在另一个树查看器(例如Archaeopteryx)中打开这个DeliverML文件,颜色也会显示在那里。
>>> import sys
>>> n = Phylo.write(tree, sys.stdout, "phyloxml")
<phyloxml ...>
<phylogeny rooted="true">
<clade>
<color>
<red>128</red>
<green>128</green>
<blue>128</blue>
</color>
<clade>
<clade>
<clade>
<name>A</name>
</clade>
<clade>
<name>B</name>
</clade>
</clade>
<clade>
<color>
<red>0</red>
<green>0</green>
<blue>255</blue>
</color>
<clade>
<name>C</name>
</clade>
...
</clade>
</phylogeny>
</phyloxml>
>>> n
1
本章的其余部分将更详细地介绍Bio.Phylo的核心功能。有关使用Bio.Phylo的更多示例,请参阅Biopython.org上的食谱页面:
I/O功能
与SeqIO和AlignIO一样,Phylo通过四个功能处理文件输入和输出: parse , read , write 和 convert ,所有这些都支持树文件格式Newick、NEXUS、SEARCH ML和NeML,以及比较数据分析Ontology(CDAO)。
的 read 函数解析给定文件中的一棵树并返回它。小心;如果文件包含多棵树或没有树,就会引发错误。
>>> from Bio import Phylo
>>> tree = Phylo.read("Tests/Nexus/int_node_labels.nwk", "newick")
>>> print(tree)
Tree(rooted=False, weight=1.0)
Clade(branch_length=75.0, name='gymnosperm')
Clade(branch_length=25.0, name='Coniferales')
Clade(branch_length=25.0)
Clade(branch_length=10.0, name='Tax+nonSci')
Clade(branch_length=90.0, name='Taxaceae')
Clade(branch_length=125.0, name='Cephalotaxus')
...
(示例文件可在 Tests/Nexus/ 和 Tests/PhyloXML/ Biopython发行版的目录。)
要处理多个(或未知数量)树,请使用 parse 函数迭代给定文件中的每个树:
>>> trees = Phylo.parse("Tests/PhyloXML/phyloxml_examples.xml", "phyloxml")
>>> for tree in trees:
... print(tree)
...
Phylogeny(description='phyloXML allows to use either a "branch_length" attribute...', name='example from Prof. Joe Felsenstein's book "Inferring Phyl...', rooted=True)
Clade()
Clade(branch_length=0.06)
Clade(branch_length=0.102, name='A')
...
将树或树的迭代写回文件 write 功能:
>>> trees = Phylo.parse("Tests/PhyloXML/phyloxml_examples.xml", "phyloxml")
>>> tree1 = next(trees)
>>> Phylo.write(tree1, "tree1.nwk", "newick")
1
>>> Phylo.write(trees, "other_trees.xml", "phyloxml") # write the remaining trees
13
使用在任何支持的格式之间转换文件 convert 功能:
>>> Phylo.convert("tree1.nwk", "newick", "tree1.xml", "nexml")
1
>>> Phylo.convert("other_trees.xml", "phyloxml", "other_trees.nex", "nexus")
13
要使用字符串而不是实际文件作为输入或输出,请使用 StringIO 就像SeqIO和AlignIO一样:
>>> from Bio import Phylo
>>> from io import StringIO
>>> handle = StringIO("(((A,B),(C,D)),(E,F,G));")
>>> tree = Phylo.read(handle, "newick")
查看和输出树木
概览的最简单方法 Tree 目的是 print 它:
>>> from Bio import Phylo
>>> tree = Phylo.read("PhyloXML/example.xml", "phyloxml")
>>> print(tree)
Phylogeny(description='phyloXML allows to use either a "branch_length" attribute...', name='example from Prof. Joe Felsenstein's book "Inferring Phyl...', rooted=True)
Clade()
Clade(branch_length=0.06)
Clade(branch_length=0.102, name='A')
Clade(branch_length=0.23, name='B')
Clade(branch_length=0.4, name='C')
这本质上是Biopython用来表示树的对象层次结构的轮廓。但更有可能的是,您想看到这棵树的图画。有三个功能可以做到这一点。
正如我们在演示中看到的那样, draw_ascii 将树的ascii艺术绘图(根字形图)打印到标准输出,或将打开的文件柄(如果给出)。并未显示有关树的所有可用信息,但它提供了一种无需依赖任何外部依赖项即可快速查看树的方法。
>>> tree = Phylo.read("PhyloXML/example.xml", "phyloxml")
>>> Phylo.draw_ascii(tree)
__________________ A
__________|
_| |___________________________________________ B
|
|___________________________________________________________________________ C
的 draw 函数使用matplotlib库绘制更有吸引力的图像。请参阅API文档,了解其接受以自定义输出的参数的详细信息。
>>> Phylo.draw(tree, branch_labels=lambda c: c.branch_length)
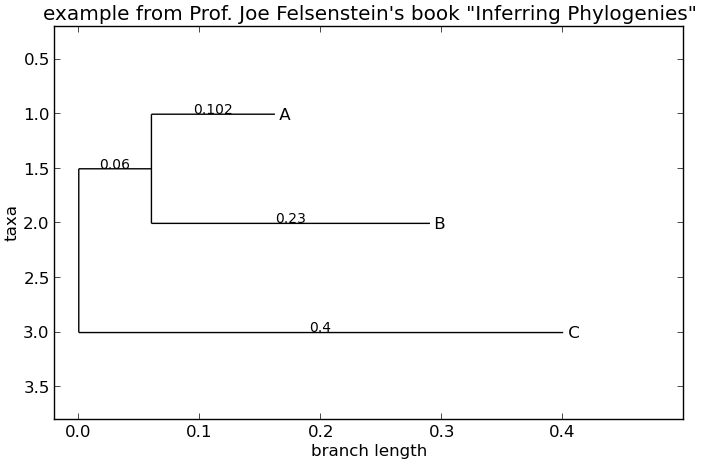
图 8 一棵简单的有根树, draw 功能
看到 图 8 比如
请参阅Biopython维基上的Phylo页面(http://biopython.org/wiki/Phylo),了解中更高级功能的描述和示例 draw_ascii , draw_graphviz 和 to_networkx .
使用Tree和Clade对象
的 Tree 制作的物品 parse 和 read 是循环子树的容器,附加到 Tree 物体 root 属性(无论系统发生树是否实际上被认为是根的)。一 Tree 已在全球范围内应用了系统发育信息,例如根性,并引用了单一的 Clade ; a Clade 具有特定于节点和分支的信息,例如分支长度以及其自己的后代列表 Clade 实例,附在 clades 属性
所以有区别 tree 和 tree.root .不过,在实践中,您很少需要担心它。为了消除差异,两者都 Tree 和 Clade 继承自 TreeMixin ,其中包含通常用于搜索、检查或修改树或其任何分支的方法的实现。这意味着几乎所有支持的方法 tree 上还提供 tree.root 以及其下方的任何进化枝。 (Clade 也有一个 root 属性,它返回分支对象本身。)
搜索和穿越方法
为了方便起见,我们提供了几种简化的方法,可以直接以列表的形式返回所有外部或内部节点:
get_terminals列出该树的所有终端(叶)节点。
get_nonterminals列出此树的所有非终端(内部)节点。
这两者都包装了一个完全控制树穿越的方法, find_clades .另外两种穿越方法, find_elements 和 find_any ,依赖相同的核心功能并接受相同的参数,由于缺乏更好的描述,我们将其称为“目标规范”。这些指定在迭代期间将匹配和返回树中的哪些对象。第一个参数可以是以下任何类型:
A TreeElement instance ,哪些树元素将根据身份匹配-因此以Clade实例作为目标进行搜索将在树中找到该Clade;
A string, which matches tree elements’ string representation — in particular, a clade’s
name(added in Biopython 1.56);A class 或 type ,其中相同类型(或子类型)的每个树元素都将被匹配;
A dictionary 其中键是树元素属性,值与每个树元素的相应属性匹配。这一个变得更加详细:
如果
int如果给定,它匹配数字上相等的属性,例如1将匹配1或1.0如果给出了布尔值(True或False),则相应的属性值将被评估为布尔值并检查是否相同
None匹配None如果给定了一个字符串,则该值被视为正则表达式(必须匹配相应元素属性中的整个字符串,而不仅仅是前缀)。一个没有特殊正则表达式字符的给定字符串将完全匹配字符串属性,所以如果你不使用正则表达式,不用担心。例如,在一个进化枝名称为Foo1,Foo2和Foo3的树中,
tree.find_clades({"name": "Foo1"})匹配Foo 1,{"name": "Foo.*"}与所有三个分支都匹配,{"name": "Foo"}与任何东西都不匹配。
由于浮点算术会产生一些奇怪的行为,因此我们不支持匹配
floatr直接。相反,使用布尔值True要匹配指定属性中具有非零值的每个元素,然后使用不等式(或精确数字,如果您喜欢危险地生活)手动过滤该属性。如果字典包含多个条目,则匹配元素必须匹配每个给定属性值-认为“和”,而不是“或”。
A function 采用单个参数(它将应用于树中的每个元素),返回True或False。为了方便起见,LookupLock、LocalMessage和ValueLock被静音,因此这提供了另一种在树中搜索浮点值或一些更复杂的特征的安全方法。
在target之后,有两个可选的关键字参数:
- 终端
— A boolean value to select for or against terminal clades (a.k.a. leaf nodes): True searches for only terminal clades, False for non-terminal (internal) clades, and the default, None, searches both terminal and non-terminal clades, as well as any tree elements lacking the
is_terminalmethod.- 秩序
— Tree traversal order:
"preorder"(default) is depth-first search,"postorder"is DFS with child nodes preceding parents, and"level"is breadth-first search.
最后,这些方法接受任意关键字参数,这些参数的处理方式与字典目标规范相同:关键字指示要搜索的元素属性的名称,参数值(字符串、integer、无或布尔值)与找到的每个属性的值进行比较。如果没有给出关键字参数,则匹配任何TreeElement类型。用于此的代码通常比传递字典作为目标规范要短: tree.find_clades({"name": "Foo1"}) 可缩短至 tree.find_clades(name="Foo1") .
(In Biopython 1.56或更高版本,这可以更短: tree.find_clades("Foo1") )
既然我们已经掌握了目标规范,以下是用于穿越树的方法:
find_clades查找包含匹配元素的每个进化枝。也就是说,找到每个元素,与
find_elements,但返回相应的进化枝对象。(This通常是您想要的。)结果可迭代所有匹配对象,默认情况下搜索深度优先。这不一定与Newick、Nexus或ML源文件中元素的出现顺序相同!
find_elements查找与给定属性匹配的所有树元素,并返回匹配的元素本身。简单的Newick树没有复杂的子元素,因此其行为与
find_clades在他们身上。PhyloML树通常确实有复杂的对象连接到分支,因此这种方法对于提取这些对象很有用。find_any返回找到的第一个元素
find_elements(),或无。这对于检查树中是否存在任何匹配元素也很有用,并且可以在条件中使用。
还有两种方法可以帮助在树中的节点之间导航:
get_path列出直接位于树根(或当前分支)和给定目标之间的分支。返回此路径上所有进化枝对象的列表,以给定目标结束,但不包括根进化枝。
trace此树中两个目标之间的所有进化枝对象的列表。不包括开始,包括结束。
信息化手段
这些方法提供有关整棵树(或任何进化枝)的信息。
common_ancestor找到所有给定目标的最近共同祖先。(This将是一个Clade对象)。如果没有给出目标,返回当前进化枝的根(调用此方法的根);如果给出了1个目标,则返回目标本身。但是,如果在当前树(或进化枝)中找不到任何指定的目标,则会引发异常。
count_terminals计算树中的终端(叶)节点的数量。
depths创建树木分支到深处的映射。结果是一个字典,其中的键是树中的所有Clade实例,值是从根到每个Clade(包括终端)的距离。默认情况下,距离是通向进化枝的累积分支长度,但与
unit_branch_lengths=True选项,仅计算分支数量(树中的级别)。distance计算两个目标之间的分支长度之和。如果仅指定了一个目标,则另一个目标就是此树的根。
total_branch_length计算此树中所有分支长度的总和。这通常被称为Inbox遗传学中树的“长度”,但我们使用更明确的名称以避免与Python术语混淆。
其余方法都是布尔检查:
is_bifurcating如果树严格分叉,则为真;即所有节点都有2个或0个子节点(分别是内部或外部)。根可能有3个后代,并且仍然被认为是分叉树的一部分。
is_monophyletic测试所有给定目标是否包括一个完整的亚进化枝-即,存在一个分支,其终端与给定目标相同。目标应该是树的终端。为了方便起见,如果目标是单系的,则此方法返回目标的共同祖先(MCRA)(而不是值
True),而且False否则。is_parent_of如果目标是此树的后代,则为真-不需要是直接后代。要检查分支的直系后代,只需使用列表成员资格测试即可:
if subclade in clade: ...is_preterminal如果所有直接后代都是终结者,则为真;如果任何直接后代都不是终结者,则为假。
改性方法
这些方法就地修改树。如果您想保持原始树完整,请首先使用Python的创建该树的完整副本 copy 模块:
tree = Phylo.read("example.xml", "phyloxml")
import copy
newtree = copy.deepcopy(tree)
collapse从树中初始化目标,将其子级重新链接到其父级。
collapse_all折叠这棵树的所有后代,只留下终端。分支长度被保留,即到每个终端的距离保持相同。对于目标规范(参见上文),仅折叠与规范匹配的内部节点。
ladderize根据终端节点的数量对分支进行就地排序。默认情况下,最深的分支位于最后。使用
reverse=True从最深到最浅对分支进行排序。prune从树上嫁接出末端进化枝。如果该分类群来自分叉,则连接节点将折叠,并将其分支长度添加到剩余的末端节点上。这可能不再是一个有意义的价值。
root_with_outgroup使用包含给定目标的外群进化支(即外群的共同祖先)重新拔出这棵树。此方法仅适用于Tree对象,而不适用于Clades。
如果外组与self.root相同,则不会发生任何更改。如果外群分支是终端(例如,给出一个终端节点作为外群),则创建一个新的分支根分支,该分支具有到给定外群的0长度分支。否则,外群底部的内部节点将成为整棵树的三叉根。如果原来的根分叉,它就会从树上掉下来。
在所有情况下,树的总分支长度保持不变。
root_at_midpoint在树的两个最远尖端之间的计算中点处重新拔出这棵树。(This使用
root_with_outgroup引擎盖下。)split生成 n (默认2)新后代。在物种树中,这是一个物种形成事件。新的分支有既定的
branch_length并且与该进化枝的根名称相同,加上一个整尾后缀(从0开始计数)-例如,分裂名为“A”的进化枝会产生子进化枝“A0”和“A1”。
有关使用可用方法的更多示例,请参阅Biopython维基上的Phylo页面(http://biopython.org/wiki/Phylo)。
PhyloHTML树的特征
Deliverable HTML文件格式包括用于注释具有其他数据类型和视觉线索的树的字段。
请参阅Biopython维基上的PhyloML页面(http://biopython.org/wiki/PhyloML),了解使用PhyloML提供的额外注释功能的描述和示例。
运行外部应用程序
虽然Bio.Phylo不会根据排列本身推断树木,但有第三方程序可以这样做。这些可以通过使用 subprocess module.
以下是如何使用Python脚本与PhyML交互的示例(http://www.atgc-montpellier.fr/phyml/)。该程序接受输入对齐 phylip-relaxed 格式(即Phylip格式,但分类单元名称没有10个字符的限制)和各种选项。
>>> import subprocess
>>> cmd = "phyml -i Tests/Phylip/random.phy"
>>> results = subprocess.run(cmd, shell=True, stdout=subprocess.PIPE, text=True)
“stdout = subProcess.PIPE”参数使程序的输出可以通过“results.stdout”访问,以用于调试目的(同样可以对“stderra”进行),并且“text=True”使返回的信息是pony字符串,而不是“bytes”对象。
这将生成一个树文件和一个具有以下名称的统计文件 [input filename] _phyml_tree.txt 和 [input filename] _phyml_stats.txt .树文件采用Newick格式:
>>> from Bio import Phylo
>>> tree = Phylo.read("Tests/Phylip/random.phy_phyml_tree.txt", "newick")
>>> Phylo.draw_ascii(tree)
__________________ F
|
| I
|
_| ________ C
| ________|
| | | , J
| | |________|
| | | , H
|___________| |__________|
| |______________ D
|
, G
|
| , E
|________________|
| ___________________________ A
|________________|
|_________ B
的 subprocess 模块还可用于与提供命令行界面的任何其他程序交互,例如RAxML(https://sco.h-its.org/exelixis/software.html)、FastTree(http://www.microbesonline.org/fasttree/)、 dnaml 和 protml .
PAML集成
Biopython 1.58支持PAML(http://abacus.gene.ucl.ac.uk/software/paml.html),这是一套按最大可能性进行系统发育分析的程序。目前已实施Codeml、baseml和yn 00程序。由于PAML使用控制文件而不是命令行参数来控制运行时选项,因此此包装器的使用偏离了Biopython中其他应用程序包装器的格式。
典型的工作流程是初始化PAML对象,指定对齐文件、树文件、输出文件和工作目录。接下来,通过 set_options() 方法或通过读取现有的控制文件。最后,程序通过 run() 方法,输出文件会自动解析为结果字典。
下面是codeml的典型用法:
>>> from Bio.Phylo.PAML import codeml
>>> cml = codeml.Codeml()
>>> cml.alignment = "Tests/PAML/Alignments/alignment.phylip"
>>> cml.tree = "Tests/PAML/Trees/species.tree"
>>> cml.out_file = "results.out"
>>> cml.working_dir = "./scratch"
>>> cml.set_options(
... seqtype=1,
... verbose=0,
... noisy=0,
... RateAncestor=0,
... model=0,
... NSsites=[0, 1, 2],
... CodonFreq=2,
... cleandata=1,
... fix_alpha=1,
... kappa=4.54006,
... )
>>> results = cml.run()
>>> ns_sites = results.get("NSsites")
>>> m0 = ns_sites.get(0)
>>> m0_params = m0.get("parameters")
>>> print(m0_params.get("omega"))
现有的输出文件也可以使用模块的 read() 功能:
>>> results = codeml.read("Tests/PAML/Results/codeml/codeml_NSsites_all.out")
>>> print(results.get("lnL max"))
这个新模块的详细文档目前位于Biopython wiki:http://biopython.org/wiki/PAML
未来计划
Bio.Phylo正在积极开发中。以下是我们可能会在未来的版本中添加的一些功能:
- 新方法
通常用于操作Tree或Clade对象的有用函数首先出现在Biopython维基上,以便临时用户可以测试它们并决定它们是否有用,然后再将它们添加到Bio。Phylo:
- Bio.Nexus端口
该模块的大部分内容是在NESCent的主持下在Google Summer of Code 2009期间编写的,作为一个实现Python支持的项目,旨在实现Python对Ottle HTML数据格式(请参阅 PhyloHTML树的特征 ).通过将现有Bio.Nexus模块的一部分移植到Bio.Phylo使用的新类,添加了对Newick和Nexus格式的支持。
目前,Bio.Nexus包含一些尚未移植到Bio.Phylo类的有用功能-特别是计算共识树。如果您发现Bio.Phylo中缺乏一些功能,请尝试查看Bio.Nexus以查看是否有它。
我们愿意接受任何改进此模块功能和可用性的建议;只需在邮件列表或我们的错误数据库中告诉我们即可。
最后,如果您需要Phylo模块中尚未包含的其他功能,请检查另一个用于蚯蚓遗传学的高质量Python库中是否可用,例如DendroPy(https://stendropy.org/)或PyCogent(http://pycogent.org/)。由于这些库还支持系统发生树的标准文件格式,因此您可以通过写入临时文件或StringIO对象来轻松地在库之间传输数据。