包括基因组图的图形
的 Bio.Graphics module depends on the third party Python library ReportLab .尽管ReportLab专注于生成PDF文件,但它还可以创建封装的PostScript(PS)和(JPEG)文件。除了这些基于载体的图像之外,还提供了某些进一步的依赖关系,例如 Python Imaging Library (PIL) 安装后,ReportLab还可以输出位图图像(包括JPEG、PNG、GIF、BMP和PICT格式)。
GenomeDiagram
介绍
的 Bio.Graphics.GenomeDiagram 模块被添加到Biopython 1.50中,之前作为独立的Python模块依赖于Biopython。Pritchard等人(2006年)在生物信息学杂志出版物中描述了基因组图。 [Pritchard2006], 其中包括一些示例图像。这里有旧手册的PDF副本,http://biopython.org/DIST/docs/GenomeDiagram/userguide.pdf,其中有更多示例。
顾名思义,GenomeDiagram旨在绘制整个基因组,特别是原核基因组,可以作为线性图(可选地分解成片段以更好地适应)或作为圆环图。看看Toth中的图2 et al. (2006) [Toth2006] 作为一个很好的例子。事实证明,它也非常适合绘制较小基因组(例如噬菌体、载体或线粒体)的相当详细的数字,例如,参见Van der Auwera中的图1和2 et al. (2009) [Vanderauwera2009] (显示带有额外的手动编辑)。
如果您将基因组加载为 SeqRecord 包含大量 SeqFeature 对象-例如从ESB文件加载(请参阅章节 顺序注释对象 和 序列输入/输出 ).
图表、轨迹、功能集和功能
GenomeDiagram使用一组嵌套的对象。在顶层,您有一个图表对象,代表沿着水平轴(或圆)的序列(或序列区域)。图表可以包含一个或多个轨道,垂直堆叠(或在圆形图表上呈放射状)。这些通常都具有相同的长度并代表相同的序列区域。您可以使用一个轨道来显示基因位置,另一个轨道来显示调节区域,第三个轨道来显示GC百分比。
最常用的曲目类型将包含捆绑在功能集中的功能。您可以选择对所有CDS功能使用一个功能集,并对TLR功能使用另一个功能集。这不是必需的--它们可以全部放入同一个特征集中,但它可以更容易地更新仅选定特征的属性(例如将所有TLR特征变成红色)。
建立完整图表有两种主要方法。首先,采用自上而下的方法,创建一个图表对象,然后使用其方法添加轨迹,并使用轨迹方法添加要素集,并使用其方法添加要素。其次,您可以单独创建各个对象(以适合您的代码的任何顺序),然后将它们组合起来。
自上而下的例子
我们将从 SeqRecord object read in from a GenBank file (see Chapter 序列输入/输出). This example uses the pPCP1 plasmid from Yersinia pestis biovar Microtus, the file is included with the Biopython unit tests under the GenBank folder, or online NC_005816.gb 来自我们的网站。
from reportlab.lib import colors
from reportlab.lib.units import cm
from Bio.Graphics import GenomeDiagram
from Bio import SeqIO
record = SeqIO.read("NC_005816.gb", "genbank")
我们使用自上而下的方法,因此在加载序列后,我们接下来创建一个空图表,然后添加一个(空)轨道,并向其中添加一个(空)特征集:
gd_diagram = GenomeDiagram.Diagram("Yersinia pestis biovar Microtus plasmid pPCP1")
gd_track_for_features = gd_diagram.new_track(1, name="Annotated Features")
gd_feature_set = gd_track_for_features.new_set()
现在有趣的部分--我们将每个基因 SeqFeature 我们的对象 SeqRecord ,并使用它在图表上生成特征。我们将把它们染成蓝色,在深蓝色和浅蓝色之间交替。
for feature in record.features:
if feature.type != "gene":
# Exclude this feature
continue
if len(gd_feature_set) % 2 == 0:
color = colors.blue
else:
color = colors.lightblue
gd_feature_set.add_feature(feature, color=color, label=True)
现在我们开始实际制作输出文件。这分为两个步骤,首先我们称之为 draw 方法,该方法使用ReportLab对象创建所有形状。然后我们称之为 write 将这些渲染为请求的文件格式的方法。请注意,您可以以多种文件格式输出:
gd_diagram.draw(
format="linear",
orientation="landscape",
pagesize="A4",
fragments=4,
start=0,
end=len(record),
)
gd_diagram.write("plasmid_linear.pdf", "PDF")
gd_diagram.write("plasmid_linear.eps", "EPS")
gd_diagram.write("plasmid_linear.svg", "SVG")
此外,只要您安装了依赖项,您还可以执行位图,例如:
gd_diagram.write("plasmid_linear.png", "PNG")
预期产量显示在 图 9 .
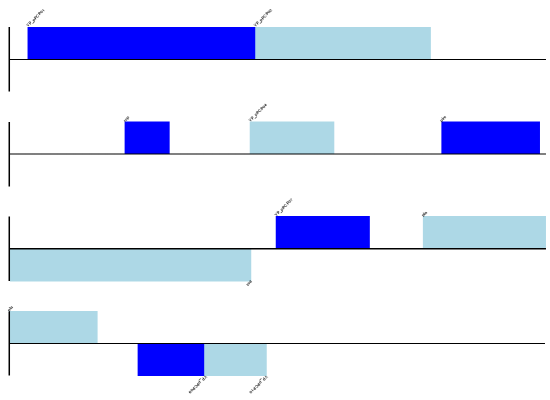
图 9 简单线性图 Y. pestis biovar Microtus pPCP 1。
注意到 fragments 我们设置为四个参数控制基因组被分解成多少个片段。
如果您想绘制圆形图形,那么尝试一下:
gd_diagram.draw(
format="circular",
circular=True,
pagesize=(20 * cm, 20 * cm),
start=0,
end=len(record),
circle_core=0.7,
)
gd_diagram.write("plasmid_circular.pdf", "PDF")
预期产量显示在 图 10 .
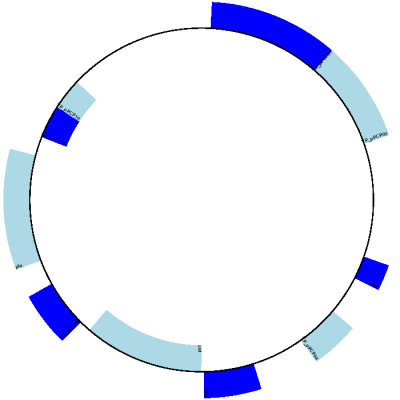
图 10 简单的圆形图 Y. pestis biovar Microtus pPCP 1。
这些数字并不是很令人兴奋,但我们才刚刚开始。
自下而上的例子
现在让我们生成完全相同的数字,但使用自下而上的方法。这意味着我们直接创建不同的对象(这几乎可以以任何顺序完成),然后将它们组合起来。
from reportlab.lib import colors
from reportlab.lib.units import cm
from Bio.Graphics import GenomeDiagram
from Bio import SeqIO
record = SeqIO.read("NC_005816.gb", "genbank")
# Create the feature set and its feature objects,
gd_feature_set = GenomeDiagram.FeatureSet()
for feature in record.features:
if feature.type != "gene":
# Exclude this feature
continue
if len(gd_feature_set) % 2 == 0:
color = colors.blue
else:
color = colors.lightblue
gd_feature_set.add_feature(feature, color=color, label=True)
# (this for loop is the same as in the previous example)
# Create a track, and a diagram
gd_track_for_features = GenomeDiagram.Track(name="Annotated Features")
gd_diagram = GenomeDiagram.Diagram("Yersinia pestis biovar Microtus plasmid pPCP1")
# Now have to glue the bits together...
gd_track_for_features.add_set(gd_feature_set)
gd_diagram.add_track(gd_track_for_features, 1)
您现在可以致电 draw 和 write 方法与之前一样使用上面自上而下示例结尾的代码来生成线性或圆形图。数字应该相同。
没有SeqPerformance的功能
在上面的例子中,我们使用了 SeqRecord ' s SeqFeature 对象来构建我们的图表(另请参阅部分 特征、位置和位置对象 ).有时你不会有 SeqFeature 对象,但只是您要绘制的特征的坐标。你必须创造最小的 SeqFeature 对象,但这很简单:
from Bio.SeqFeature import SeqFeature, SimpleLocation
my_seq_feature = SeqFeature(SimpleLocation(50, 100, strand=+1))
对于链,使用 +1 对于向前链, -1 对于反向链,并且 None 对双方来说。这是一个简短的独立示例:
from Bio.SeqFeature import SeqFeature, SimpleLocation
from Bio.Graphics import GenomeDiagram
from reportlab.lib.units import cm
gdd = GenomeDiagram.Diagram("Test Diagram")
gdt_features = gdd.new_track(1, greytrack=False)
gds_features = gdt_features.new_set()
# Add three features to show the strand options,
feature = SeqFeature(SimpleLocation(25, 125, strand=+1))
gds_features.add_feature(feature, name="Forward", label=True)
feature = SeqFeature(SimpleLocation(150, 250, strand=None))
gds_features.add_feature(feature, name="Strandless", label=True)
feature = SeqFeature(SimpleLocation(275, 375, strand=-1))
gds_features.add_feature(feature, name="Reverse", label=True)
gdd.draw(format="linear", pagesize=(15 * cm, 4 * cm), fragments=1, start=0, end=400)
gdd.write("GD_labels_default.pdf", "pdf")
输出显示在 图 11 (in默认特征颜色,淡绿色)。
请注意,我们使用了 name 此处的参数指定这些功能的标题文本。接下来将更详细地讨论这一点。
功能说明
回想一下,我们使用了以下方法(其中 feature 是一个 SeqFeature 对象)向图表添加功能:
gd_feature_set.add_feature(feature, color=color, label=True)
在上面的例子中 SeqFeature 注释用于为功能选择合理的标题。默认情况下, SeqFeature 使用对象的限定符字典: gene , label , name , locus_tag ,而且 product .更简单地,您可以直接指定名称:
gd_feature_set.add_feature(feature, color=color, label=True, name="My Gene")
除了每个要素标签的标题文本外,您还可以选择字体、位置(默认为符号的开始,您还可以选择中间或结束)和方向(仅适用于线性图,默认为旋转 \(45\) 度):
# Large font, parallel with the track
gd_feature_set.add_feature(
feature, label=True, color="green", label_size=25, label_angle=0
)
# Very small font, perpendicular to the track (towards it)
gd_feature_set.add_feature(
feature,
label=True,
color="purple",
label_position="end",
label_size=4,
label_angle=90,
)
# Small font, perpendicular to the track (away from it)
gd_feature_set.add_feature(
feature,
label=True,
color="blue",
label_position="middle",
label_size=6,
label_angle=-90,
)
将这三个片段中的每一个片段与上一节中的完整示例相结合,应该会得到类似于中的曲目的东西 图 11 .
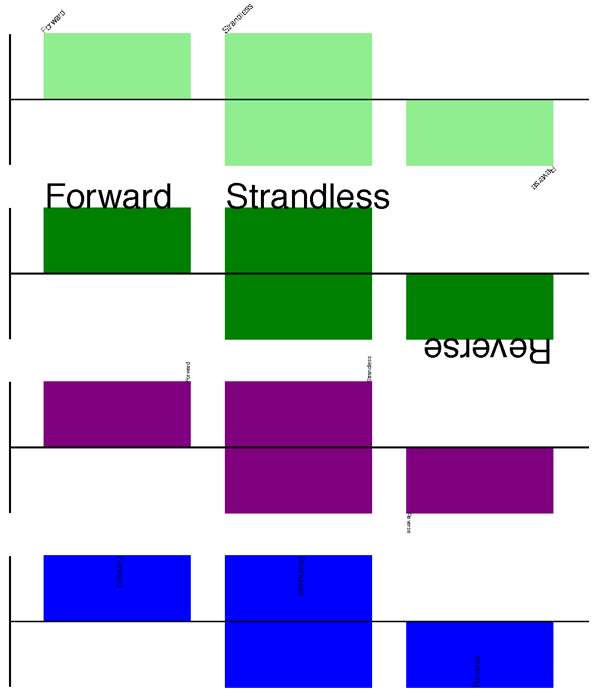
图 11 显示标签选项的简单基因组图。
顶部的淡绿色图显示默认标签设置(请参阅第节 没有SeqPerformance的功能 )而其余则显示标签大小、位置和方向的变化(请参阅第节 功能说明 ).
我们这里没有展示,但您也可以设置 label_color 控制标签的颜色(用于第部分 一个很好的例子 ).
您会注意到默认字体相当小-这是有道理的,因为您通常会在页面上绘制许多(小)特征,而不仅仅是此处显示的几个大特征。
特征标志
上面的例子都只是使用了该功能的默认标志,即普通框,这是GenomeDiagram上一次公开发布的独立版本中所提供的全部功能。当GenomeDiagram添加到Biopython 1.50时,就包含了箭头符号:
# Default uses a BOX sigil
gd_feature_set.add_feature(feature)
# You can make this explicit:
gd_feature_set.add_feature(feature, sigil="BOX")
# Or opt for an arrow:
gd_feature_set.add_feature(feature, sigil="ARROW")
Biopython 1.61又增加了三个印记,
# Box with corners cut off (making it an octagon)
gd_feature_set.add_feature(feature, sigil="OCTO")
# Box with jagged edges (useful for showing breaks in contains)
gd_feature_set.add_feature(feature, sigil="JAGGY")
# Arrow which spans the axis with strand used only for direction
gd_feature_set.add_feature(feature, sigil="BIGARROW")
这些显示在 图 12 .大多数图案都适合边界框(如默认BOX图案所示),对于正向或反向链,可以在轴上方或下方,或者对于无链特征,可以跨在轴上方(两倍高度)。BIGARROW徽标有所不同,总是横跨在从特征支架上取的方向的轴上。

图 12 简单的基因组图显示不同的印记。
箭头印记
我们在上一节中介绍了箭头印记。还有两个额外的选项来调整箭头的形状,首先是箭杆的厚度,以边界框高度的比例给出:
# Full height shafts, giving pointed boxes:
gd_feature_set.add_feature(feature, sigil="ARROW", color="brown", arrowshaft_height=1.0)
# Or, thin shafts:
gd_feature_set.add_feature(feature, sigil="ARROW", color="teal", arrowshaft_height=0.2)
# Or, very thin shafts:
gd_feature_set.add_feature(
feature, sigil="ARROW", color="darkgreen", arrowshaft_height=0.1
)
结果示于 图 13 .
图 13 简单的基因组图显示箭头轴选项。
其次,箭头的长度-作为边界框高度的比例给出(默认为 \(0.5\) ,或者 \(50\%\) ):
# Short arrow heads:
gd_feature_set.add_feature(feature, sigil="ARROW", color="blue", arrowhead_length=0.25)
# Or, longer arrow heads:
gd_feature_set.add_feature(feature, sigil="ARROW", color="orange", arrowhead_length=1)
# Or, very very long arrow heads (i.e. all head, no shaft, so triangles):
gd_feature_set.add_feature(feature, sigil="ARROW", color="red", arrowhead_length=10000)
结果示于 图 14 .
图 14 简单的基因组图显示箭头选项。
Biopython 1.61添加了一个新的 BIGARROW 始终跨在轴上的符号,反向链指向左,否则指向右:
# A large arrow straddling the axis:
gd_feature_set.add_feature(feature, sigil="BIGARROW")
上面显示的所有轴和箭头选项 ARROW 标志可以用于 BIGARROW 也有印记。
一个很好的例子
现在让我们回到pPCP 1载体 Yersinia pestis biovar Microtus ,以及部分中使用的自上而下的方法 自上而下的例子 ,但是要利用我们现在讨论的符号选项。这一次,我们将使用箭头表示基因,并用无链特征(如普通框)覆盖它们,显示一些限制性消化位点的位置。
from reportlab.lib import colors
from reportlab.lib.units import cm
from Bio.Graphics import GenomeDiagram
from Bio import SeqIO
from Bio.SeqFeature import SeqFeature, SimpleLocation
record = SeqIO.read("NC_005816.gb", "genbank")
gd_diagram = GenomeDiagram.Diagram(record.id)
gd_track_for_features = gd_diagram.new_track(1, name="Annotated Features")
gd_feature_set = gd_track_for_features.new_set()
for feature in record.features:
if feature.type != "gene":
# Exclude this feature
continue
if len(gd_feature_set) % 2 == 0:
color = colors.blue
else:
color = colors.lightblue
gd_feature_set.add_feature(
feature, sigil="ARROW", color=color, label=True, label_size=14, label_angle=0
)
# I want to include some strandless features, so for an example
# will use EcoRI recognition sites etc.
for site, name, color in [
("GAATTC", "EcoRI", colors.green),
("CCCGGG", "SmaI", colors.orange),
("AAGCTT", "HindIII", colors.red),
("GGATCC", "BamHI", colors.purple),
]:
index = 0
while True:
index = record.seq.find(site, start=index)
if index == -1:
break
feature = SeqFeature(SimpleLocation(index, index + len(site)))
gd_feature_set.add_feature(
feature,
color=color,
name=name,
label=True,
label_size=10,
label_color=color,
)
index += len(site)
gd_diagram.draw(format="linear", pagesize="A4", fragments=4, start=0, end=len(record))
gd_diagram.write("plasmid_linear_nice.pdf", "PDF")
gd_diagram.write("plasmid_linear_nice.eps", "EPS")
gd_diagram.write("plasmid_linear_nice.svg", "SVG")
gd_diagram.draw(
format="circular",
circular=True,
pagesize=(20 * cm, 20 * cm),
start=0,
end=len(record),
circle_core=0.5,
)
gd_diagram.write("plasmid_circular_nice.pdf", "PDF")
gd_diagram.write("plasmid_circular_nice.eps", "EPS")
gd_diagram.write("plasmid_circular_nice.svg", "SVG")
预期产出见图 显示选定限制性消化位点的载体pCPC 1线性图。 和 显示选定限制性消化位点的载体pCPC 1的圆形图。 .
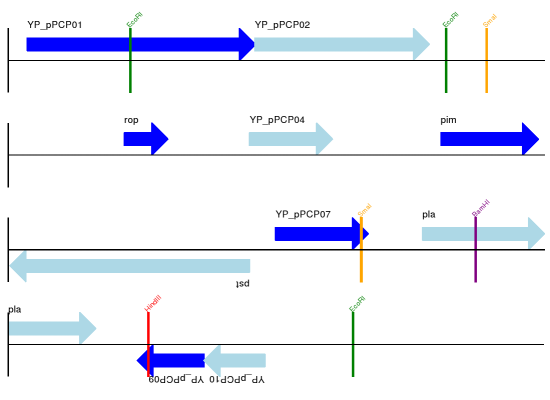
图 15 显示选定限制性消化位点的载体pCPC 1线性图。
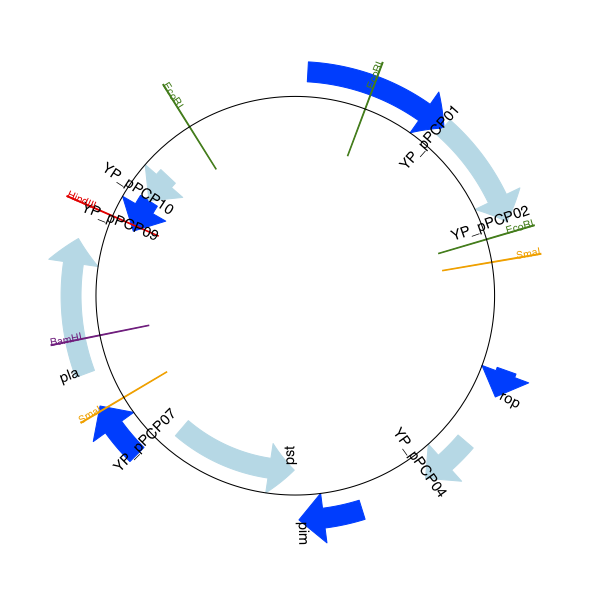
图 16 显示选定限制性消化位点的载体pCPC 1的圆形图。
多个轨道
到目前为止,所有的例子都使用了一个轨道,但是你可以有多个轨道-例如在一个轨道上显示基因,在另一个轨道上显示重复区域。在这个例子中,我们将展示三个噬菌体基因组并排的比例,灵感来自Proux的图6 et al. (2002) [Proux2002]. 我们需要以下三种噬菌体的基因库文件:
NC_002703- 乳球菌噬菌体Tuc2009,完整基因组 (\(38347\) BP)AF323668- 噬菌体bIL 285,完整基因组 (\(35538\) BP)NC_003212– Listeria innocua Clip 11262,完整基因组,我们仅关注整合的前噬菌5(长度相似)。
如果您愿意,您可以使用Deliverz下载这些内容,请参阅部分 EFetch:从Deliverz下载完整记录 了解更多详细信息。对于第三条记录,我们已经确定了噬菌体整合到基因组中的位置,并对记录进行切片以提取它(保留特征,请参阅第节 切片SeqRecord ),并且还必须反向互补以匹配前两个噬菌体的方向(再次保留特征,参见第节 反向补充SeqRecord对象 ):
from Bio import SeqIO
A_rec = SeqIO.read("NC_002703.gbk", "gb")
B_rec = SeqIO.read("AF323668.gbk", "gb")
C_rec = SeqIO.read("NC_003212.gbk", "gb")[2587879:2625807].reverse_complement(name=True)
我们正在模仿的人物使用不同的颜色来实现不同的基因功能。实现此目标的一种方法是编辑SEN文件以记录每个要素的颜色首选项-某种东西 Sanger’s Artemis editor 确实如此,GenomeDiagram应该理解这一点。然而,在这里,我们只对三个颜色列表进行硬编码。
请注意,SEN文件中的注释与Proux中显示的不完全匹配 et al. ,他们画了一些未注释的基因。
from reportlab.lib.colors import (
red,
grey,
orange,
green,
brown,
blue,
lightblue,
purple,
)
A_colors = (
[red] * 5
+ [grey] * 7
+ [orange] * 2
+ [grey] * 2
+ [orange]
+ [grey] * 11
+ [green] * 4
+ [grey]
+ [green] * 2
+ [grey, green]
+ [brown] * 5
+ [blue] * 4
+ [lightblue] * 5
+ [grey, lightblue]
+ [purple] * 2
+ [grey]
)
B_colors = (
[red] * 6
+ [grey] * 8
+ [orange] * 2
+ [grey]
+ [orange]
+ [grey] * 21
+ [green] * 5
+ [grey]
+ [brown] * 4
+ [blue] * 3
+ [lightblue] * 3
+ [grey] * 5
+ [purple] * 2
)
C_colors = (
[grey] * 30
+ [green] * 5
+ [brown] * 4
+ [blue] * 2
+ [grey, blue]
+ [lightblue] * 2
+ [grey] * 5
)
现在来绘制它们--这次我们将三个轨道添加到图表中,并注意它们被赋予不同的开始/结束值以反映其不同的长度(这需要Biopython 1.59或更高版本)。
from Bio.Graphics import GenomeDiagram
name = "Proux Fig 6"
gd_diagram = GenomeDiagram.Diagram(name)
max_len = 0
for record, gene_colors in zip([A_rec, B_rec, C_rec], [A_colors, B_colors, C_colors]):
max_len = max(max_len, len(record))
gd_track_for_features = gd_diagram.new_track(
1, name=record.name, greytrack=True, start=0, end=len(record)
)
gd_feature_set = gd_track_for_features.new_set()
i = 0
for feature in record.features:
if feature.type != "gene":
# Exclude this feature
continue
gd_feature_set.add_feature(
feature,
sigil="ARROW",
color=gene_colors[i],
label=True,
name=str(i + 1),
label_position="start",
label_size=6,
label_angle=0,
)
i += 1
gd_diagram.draw(format="linear", pagesize="A4", fragments=1, start=0, end=max_len)
gd_diagram.write(name + ".pdf", "PDF")
gd_diagram.write(name + ".eps", "EPS")
gd_diagram.write(name + ".svg", "SVG")
预期产量显示在 图 17 .
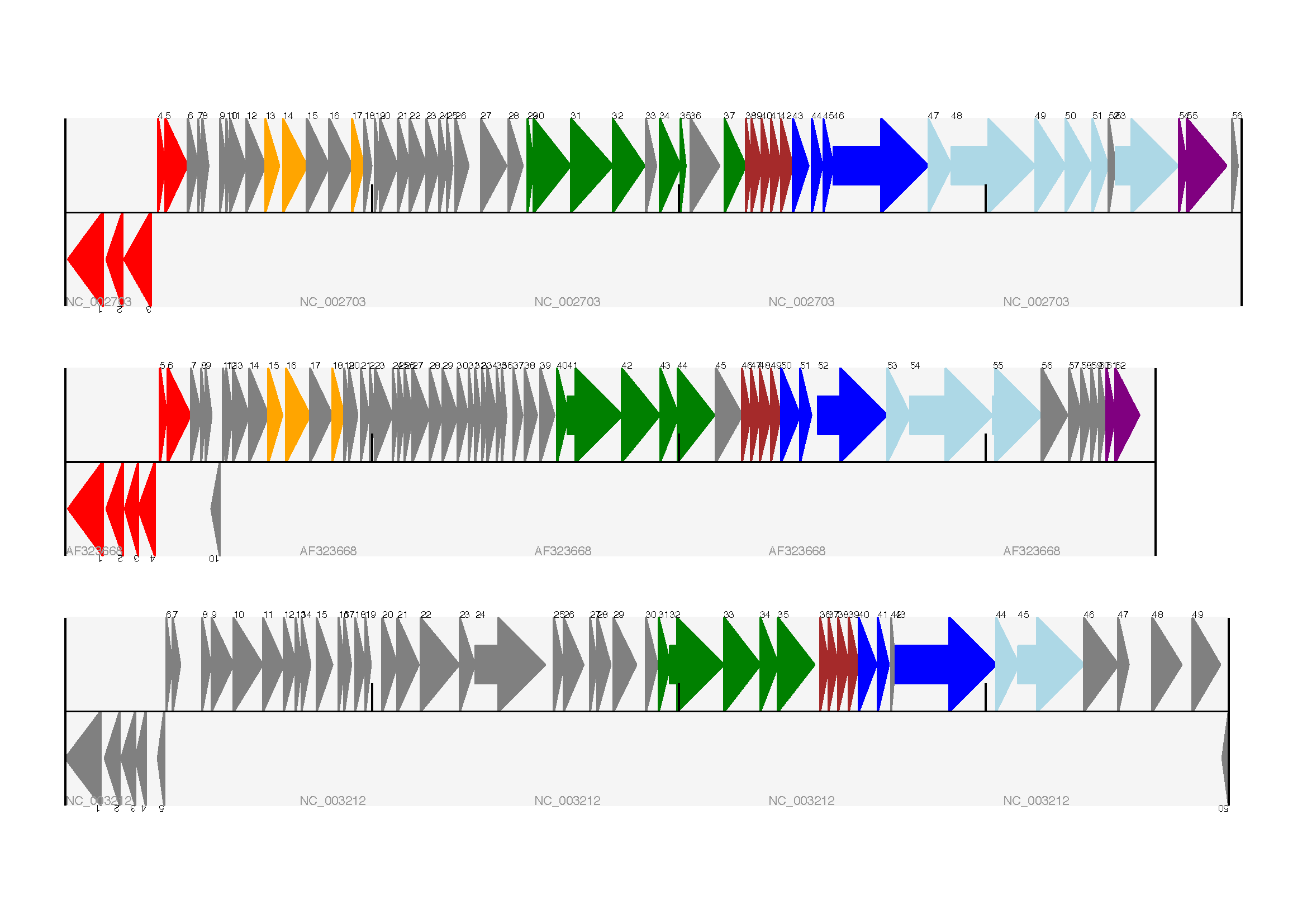
图 17 三个打印机的三个轨道的线性图。
这显示了乳球菌噬菌体Tuc2009(NC_002703)、噬菌体bIL 285(AF 323668)和来自的噬菌体5 Listeria innocua Clip 11262(NC_003212)(请参阅部分 多个轨道 ).
我确实想知道为什么在原始手稿中底部噬菌体中没有标记红色或橙色基因。另一个重要的一点是,这里显示的噬菌体具有不同的长度-这是因为它们都被绘制成相同的规模(它们 are 不同长度)。
与已发布的图的关键区别在于它们在相似蛋白质之间具有颜色编码的链接-这就是我们将在下一节中做的事情。
轨道之间的交叉链接
Biopython 1.59添加了在轨道之间绘制交叉链接的功能--既有简单的线性图,我们将在这里展示,也有分解为碎片和圆形图的线性图。
继续上一节的示例,其灵感来自Proux的图6 et al. 2002 [Proux2002], 我们需要一份基因对之间交叉链接的列表,以及要使用的分数或颜色。实际上,您可能会通过计算从AMPS文件中提取它,但在这里我手动输入了它们。
我的命名惯例继续将这三个噬菌体称为A、B和C。下面是我们想要显示的A和B之间的链接,以元组列表的形式给出(相似性分数百分比,A中的基因,B中的基因)。
# Tuc2009 (NC_002703) vs bIL285 (AF323668)
A_vs_B = [
(99, "Tuc2009_01", "int"),
(33, "Tuc2009_03", "orf4"),
(94, "Tuc2009_05", "orf6"),
(100, "Tuc2009_06", "orf7"),
(97, "Tuc2009_07", "orf8"),
(98, "Tuc2009_08", "orf9"),
(98, "Tuc2009_09", "orf10"),
(100, "Tuc2009_10", "orf12"),
(100, "Tuc2009_11", "orf13"),
(94, "Tuc2009_12", "orf14"),
(87, "Tuc2009_13", "orf15"),
(94, "Tuc2009_14", "orf16"),
(94, "Tuc2009_15", "orf17"),
(88, "Tuc2009_17", "rusA"),
(91, "Tuc2009_18", "orf20"),
(93, "Tuc2009_19", "orf22"),
(71, "Tuc2009_20", "orf23"),
(51, "Tuc2009_22", "orf27"),
(97, "Tuc2009_23", "orf28"),
(88, "Tuc2009_24", "orf29"),
(26, "Tuc2009_26", "orf38"),
(19, "Tuc2009_46", "orf52"),
(77, "Tuc2009_48", "orf54"),
(91, "Tuc2009_49", "orf55"),
(95, "Tuc2009_52", "orf60"),
]
B和C也是如此:
# bIL285 (AF323668) vs Listeria innocua prophage 5 (in NC_003212)
B_vs_C = [
(42, "orf39", "lin2581"),
(31, "orf40", "lin2580"),
(49, "orf41", "lin2579"), # terL
(54, "orf42", "lin2578"), # portal
(55, "orf43", "lin2577"), # protease
(33, "orf44", "lin2576"), # mhp
(51, "orf46", "lin2575"),
(33, "orf47", "lin2574"),
(40, "orf48", "lin2573"),
(25, "orf49", "lin2572"),
(50, "orf50", "lin2571"),
(48, "orf51", "lin2570"),
(24, "orf52", "lin2568"),
(30, "orf53", "lin2567"),
(28, "orf54", "lin2566"),
]
对于第一个和最后一个噬菌体,这些标识符是位点标签,对于中间的噬菌体,没有位点标签,所以我用基因名称代替。下面的小助手函数允许我们使用基因座标签或基因名称查找特征:
def get_feature(features, id, tags=["locus_tag", "gene"]):
"""Search list of SeqFeature objects for an identifier under the given tags."""
for f in features:
for key in tags:
# tag may not be present in this feature
for x in f.qualifiers.get(key, []):
if x == id:
return f
raise KeyError(id)
我们现在可以将这些标识符对列表转换为SeqFeature对,从而找到它们的位置坐标。我们现在可以将所有这些代码和下面的代码片段添加到前面的示例中(就在 gd_diagram.draw(...) line – see the finished example script Proux_et_al_2002_Figure_6.py 纳入 Doc/examples Biopython源代码的文件夹)将交叉链接添加到图中:
from Bio.Graphics.GenomeDiagram import CrossLink
from reportlab.lib import colors
# Note it might have been clearer to assign the track numbers explicitly...
for rec_X, tn_X, rec_Y, tn_Y, X_vs_Y in [
(A_rec, 3, B_rec, 2, A_vs_B),
(B_rec, 2, C_rec, 1, B_vs_C),
]:
track_X = gd_diagram.tracks[tn_X]
track_Y = gd_diagram.tracks[tn_Y]
for score, id_X, id_Y in X_vs_Y:
feature_X = get_feature(rec_X.features, id_X)
feature_Y = get_feature(rec_Y.features, id_Y)
color = colors.linearlyInterpolatedColor(
colors.white, colors.firebrick, 0, 100, score
)
link_xy = CrossLink(
(track_X, feature_X.location.start, feature_X.location.end),
(track_Y, feature_Y.location.start, feature_Y.location.end),
color,
colors.lightgrey,
)
gd_diagram.cross_track_links.append(link_xy)
此代码有几个重要的部分。首先 GenomeDiagram 对象具有 cross_track_links 属性,它只是一个列表 CrossLink 对象每个 CrossLink 对象采用两组特定于轨道的坐标(这里以二元组形式给出,您可以选择使用 GenomeDiagram.Feature 相反,对象)。您可以选择提供颜色、边框颜色,并说明是否应该翻转绘制此链接(对于显示翻转很有用)。
您还可以看到我们如何将BST百分比同一性分数转化为颜色,并在白色之间进行插值 (\(0\%\) )和深红色 (\(100\%\) ).在这个例子中,我们没有任何交叉链接重叠的问题。解决这个问题的一种方法是在ReportLab中使用透明度,通过使用具有Alpha通道集的颜色。然而,这种着色配色方案与重叠透明度相结合将很难解释。预期产量显示在 图 18 .
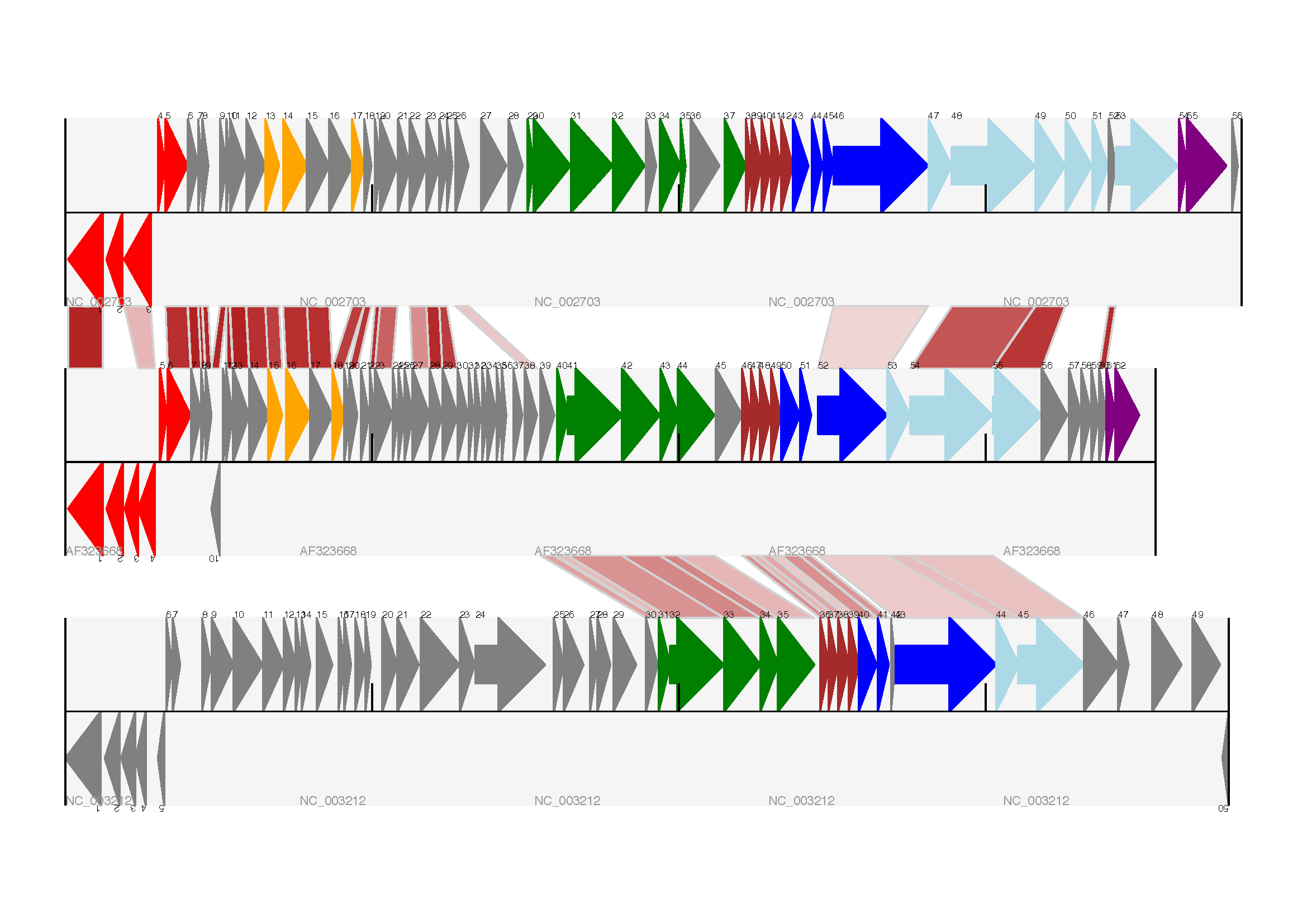
图 18 具有三个轨道加上基本交叉链接的线性图。
这三条轨迹显示乳球菌噬菌体Tuc2009(NC_002703)、噬菌体bIL 285(AF 323668)和来自 Listeria innocua Clip 11262(NC_003212)加上用百分比同一性着色的基本交叉链接(请参阅部分 轨道之间的交叉链接 ).
Biopython内还有很多工作可以帮助改善这一数字。首先,这种情况下的交叉连接是在特定链中绘制的蛋白质之间的。它可以帮助在要素轨道上添加背景区域(使用“BOX”符号的要素)以扩展交叉链接。此外,我们可以降低要素轨迹的垂直高度,以便为链接分配更多内容-一种方法是为空轨迹分配空间。此外,在这种没有大基因重叠的情况下,我们可以使用轴跨 BIGARROW sigil, which allows us to further reduce the vertical space needed for the track. These improvements are demonstrated in the example script Proux_et_al_2002_Figure_6.py 纳入 Doc/examples Biopython源代码的文件夹。预期产量显示在 图 19 .
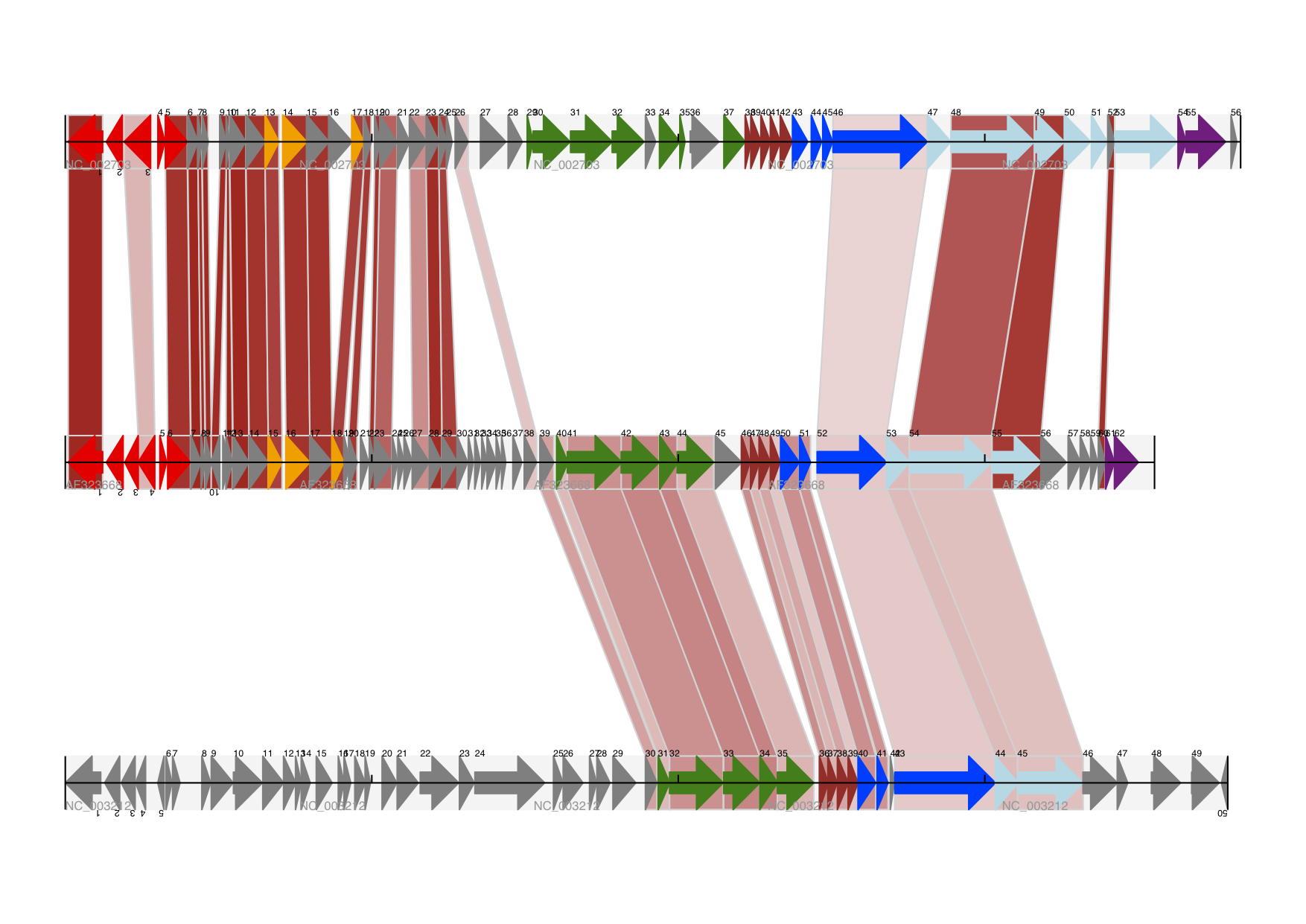
图 19 具有三个轨道和阴影交叉链接的线性图。
这三条轨迹显示乳球菌噬菌体Tuc2009(NC_002703)、噬菌体bIL 285(AF 323668)和来自 Listeria innocua Clip 11262(NC_003212)加上交叉链接(用百分比同一性阴影)(请参阅第节 轨道之间的交叉链接 ).
除此之外,您可能希望在矢量图像编辑器中手动完成的收尾工作包括微调基因标签的位置,以及添加其他自定义注释,例如突出显示特定区域。
尽管在本例中并没有真正必要,因为没有交叉链接重叠,但在ReportLab中使用透明颜色对于叠加多个链接来说是一种非常有用的技术。然而,在这种情况下,应避免阴影配色方案。
进一步的选项
您可以控制勾号来显示比例-毕竟每个图表都应该显示其单位以及灰色轨道标签的数量。
此外,我们只使用了 FeatureSet 迄今基因组图还有一个 GraphSet 其可用于显示线性图、条形图和热图(例如,显示平行于特征的轨道上的GC%图)。
此处尚未涵盖这些选项,因此目前我们将您推荐至 User Guide (PDF) 包含在GenomeDiagram的独立版本中(但请先阅读下一部分)和文档字符串中。
转换旧代码
如果您有使用GenomeDiagram独立版本编写的旧代码,并且您想将其切换到使用Biopython包含的新版本,那么您将必须进行一些更改-最重要的是对您的导入声明。
此外,旧版本的GenomeDiagram仅使用英国的颜色和中心拼写(colour和中心)。您需要更改为美国拼写,尽管几年来Biopython版本的GenomeDiagram支持这两种拼写。
例如,如果您曾经拥有:
from GenomeDiagram import GDFeatureSet, GDDiagram
gdd = GDDiagram("An example")
...
您可以像这样切换导入声明:
from Bio.Graphics.GenomeDiagram import FeatureSet as GDFeatureSet, Diagram as GDDiagram
gdd = GDDiagram("An example")
...
希望这就足够了。从长远来看,您可能想要切换到新名称,但您必须更改更多代码:
from Bio.Graphics.GenomeDiagram import FeatureSet, Diagram
gdd = Diagram("An example")
...
或者:
from Bio.Graphics import GenomeDiagram
gdd = GenomeDiagram.Diagram("An example")
...
如果您遇到困难,请在Biopython邮件列表中询问建议。一个问题是我们没有包含旧模块 GenomeDiagram.GDUtilities 呢这包括许多GC%相关功能,这些功能可能会合并到 Bio.SeqUtils 稍后
染色体
的 Bio.Graphics.BasicChromosome 模块允许绘制染色体。珍妮有一个例子 et al. (2012) [Jupe2012] (open访问)使用颜色来突出不同的基因家族。
简单染色体
这是一个非常简单的例子-我们将使用 Arabidopsis thaliana .

图 20 简单的染色体图 Arabidopsis thaliana .
您可以跳过这一点,但首先我从NCBI的RTP网站ftp://ftp.ncbi.nlm.nih.gov/genomes/archive/old_refseq/Arabidopsis_thaliana/下载了五条测序染色体作为五个单独的FASTA文件,然后用 Bio.SeqIO 以找出它们的长度。您可以为此使用ESB文件(下一个示例使用这些文件来绘制特征),但如果您想要的只是长度,那么对整个染色体使用FASTA文件会更快:
from Bio import SeqIO
entries = [
("Chr I", "CHR_I/NC_003070.fna"),
("Chr II", "CHR_II/NC_003071.fna"),
("Chr III", "CHR_III/NC_003074.fna"),
("Chr IV", "CHR_IV/NC_003075.fna"),
("Chr V", "CHR_V/NC_003076.fna"),
]
for name, filename in entries:
record = SeqIO.read(filename, "fasta")
print(name, len(record))
这给出了五条染色体的长度,我们现在将在下面简短的演示中使用它 BasicChromosome 模块:
from reportlab.lib.units import cm
from Bio.Graphics import BasicChromosome
entries = [
("Chr I", 30432563),
("Chr II", 19705359),
("Chr III", 23470805),
("Chr IV", 18585042),
("Chr V", 26992728),
]
max_len = 30432563 # Could compute this from the entries dict
telomere_length = 1000000 # For illustration
chr_diagram = BasicChromosome.Organism()
chr_diagram.page_size = (29.7 * cm, 21 * cm) # A4 landscape
for name, length in entries:
cur_chromosome = BasicChromosome.Chromosome(name)
# Set the scale to the MAXIMUM length plus the two telomeres in bp,
# want the same scale used on all five chromosomes so they can be
# compared to each other
cur_chromosome.scale_num = max_len + 2 * telomere_length
# Add an opening telomere
start = BasicChromosome.TelomereSegment()
start.scale = telomere_length
cur_chromosome.add(start)
# Add a body - using bp as the scale length here.
body = BasicChromosome.ChromosomeSegment()
body.scale = length
cur_chromosome.add(body)
# Add a closing telomere
end = BasicChromosome.TelomereSegment(inverted=True)
end.scale = telomere_length
cur_chromosome.add(end)
# This chromosome is done
chr_diagram.add(cur_chromosome)
chr_diagram.draw("simple_chrom.pdf", "Arabidopsis thaliana")
这应该创建一个非常简单的PDF文件,如所示 图 20 .这个例子故意简短而甜蜜。下一个示例显示感兴趣要素的位置。
注释染色体
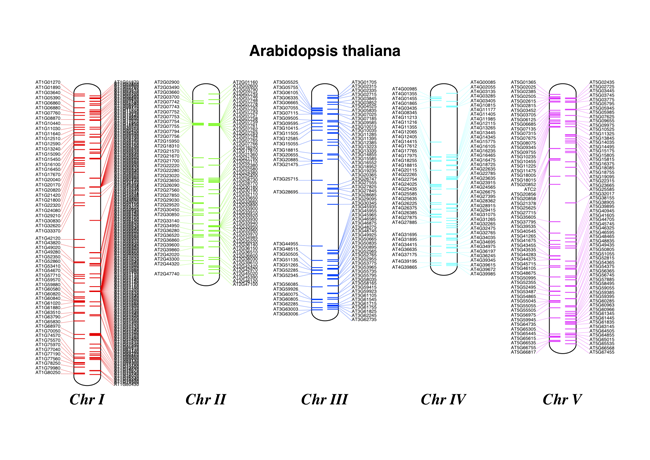
图 21 染色体图 Arabidopsis thaliana 显示TLR基因。
继续上一个例子,让我们还展示TLR基因。我们将通过解析五个人的基因库文件来获取他们的位置 Arabidopsis thaliana 染色体您需要从NCBI RTP网站ftp://ftp.ncbi.nlm.nih.gov/genomes/archive/old_refseq/Deliveropsis_thaliana/下载这些文件,并保留收件箱名称或编辑下面的路径:
from reportlab.lib.units import cm
from Bio import SeqIO
from Bio.Graphics import BasicChromosome
entries = [
("Chr I", "CHR_I/NC_003070.gbk"),
("Chr II", "CHR_II/NC_003071.gbk"),
("Chr III", "CHR_III/NC_003074.gbk"),
("Chr IV", "CHR_IV/NC_003075.gbk"),
("Chr V", "CHR_V/NC_003076.gbk"),
]
max_len = 30432563 # Could compute this from the entries dict
telomere_length = 1000000 # For illustration
chr_diagram = BasicChromosome.Organism()
chr_diagram.page_size = (29.7 * cm, 21 * cm) # A4 landscape
for index, (name, filename) in enumerate(entries):
record = SeqIO.read(filename, "genbank")
length = len(record)
features = [f for f in record.features if f.type == "tRNA"]
# Record an Artemis style integer color in the feature's qualifiers,
# 1 = Black, 2 = Red, 3 = Green, 4 = blue, 5 =cyan, 6 = purple
for f in features:
f.qualifiers["color"] = [index + 2]
cur_chromosome = BasicChromosome.Chromosome(name)
# Set the scale to the MAXIMUM length plus the two telomeres in bp,
# want the same scale used on all five chromosomes so they can be
# compared to each other
cur_chromosome.scale_num = max_len + 2 * telomere_length
# Add an opening telomere
start = BasicChromosome.TelomereSegment()
start.scale = telomere_length
cur_chromosome.add(start)
# Add a body - again using bp as the scale length here.
body = BasicChromosome.AnnotatedChromosomeSegment(length, features)
body.scale = length
cur_chromosome.add(body)
# Add a closing telomere
end = BasicChromosome.TelomereSegment(inverted=True)
end.scale = telomere_length
cur_chromosome.add(end)
# This chromosome is done
chr_diagram.add(cur_chromosome)
chr_diagram.draw("tRNA_chrom.pdf", "Arabidopsis thaliana")
它可能会警告您标签太靠近-看看Chr I的正向链(右侧),但它应该创建一个彩色PDF文件,如 图 21 .