爆炸(新)
嘿,每个人都喜欢BRAST吧?我的意思是,天哪,如何更容易地在你的一个序列与已知世界中的所有其他序列之间进行比较呢?但是,当然,这一部分并不是关于AMPS有多酷,因为我们已经知道这一点。这是关于BST的问题--处理大规模运行产生的大量数据以及一般来说自动化BST运行可能确实很困难。
幸运的是,Biopython的人们非常了解这一点,所以他们开发了很多工具来处理AMPS并让事情变得更容易。本节详细介绍了如何使用这些工具并使用它们做有用的事情。
处理AMPS可以分为两个步骤,这两个步骤都可以在Biopython内完成。首先,对您的查询序列运行BST,并获得一些输出。其次,在Python中解析BST输出以进行进一步分析。
您对运行BLAST的第一次介绍可能是通过 NCBI BLAST web page .事实上,有很多种方法可以运行AMPS,这些方法可以分为多种方式。最重要的区别是在本地运行BST(在您自己的机器上)和远程运行BST(在另一台机器上,通常是NCBI服务器)。我们将通过从Python脚本中调用NCBI在线BST服务来开始本章。
在互联网上运行AMPS
我们使用该功能 qblast 在 Bio.Blast 调用在线版本的AMPS模块。
的 NCBI guidelines 国家:
联系服务器的频率不要超过每10秒一次。
对任何单个DID进行民意调查的频率不要超过每分钟一次。
使用URL参数电子邮件和工具,以便NCBI可以在出现问题时与您联系。
如果提交超过50个搜索,则在周末或工作日东部时间晚上9点至凌晨5点之间运行脚本。
Blast.qblast 自动遵循前两点。要满足第三点,请设置 Blast.email 变量( Blast.tool 变量已设置为 "biopython" 默认情况下):
>>> from Bio import Blast
>>> Blast.tool
'biopython'
>>> Blast.email = "A.N.Other@example.com"
爆炸性论点
的 qblast 函数有三个非可选参数:
第一个参数是用于搜索的BST程序,作为大小写字符串。这些程序及其选项在 NCBI BLAST web page .目前
qblast仅适用于blastup、blastp、blastx、tblast和tblastx。第二个参数指定要搜索的数据库。同样,此选项可在 NCBI’s BLAST Help pages .
第三个参数是包含查询序列的字符串。这可以是序列本身、fasta格式的序列,也可以是GI号等标识符。
的 qblast 函数还接受许多其他选项参数,这些参数基本上类似于您可以在AMPS网页上设置的不同参数。我们将在这里重点介绍其中一些:
的论点
url_base设置在互联网上运行BST的基本URL。默认情况下,它连接到NCBI。的
qblast函数可以以各种格式返回BST结果,您可以通过可选的format_type关键字:"XML","HTML","Text","XML2","JSON2",或者"Tabular".默认值为"XML",因为这是解析器期望的格式,如部分所述 解析BST输出 下面的论点
expect设置期望或e值阈值。
有关可选的BST参数的更多信息,请参阅NCBI自己的文档或Biopython中内置的文档:
>>> from Bio import Blast
>>> help(Blast.qblast)
请注意,NCBI BST网站上的默认设置与QBST上的默认设置不完全相同。如果得到不同的结果,您需要检查参数(例如,期望值阈值和差距值)。
例如,如果您有一个核苷酸序列,您想使用BLACKS在核苷酸数据库(nt)中搜索,并且您知道查询序列的GI编号,则可以用途:
>>> from Bio import Blast
>>> result_stream = Blast.qblast("blastn", "nt", "8332116")
或者,如果我们的查询序列已经在FASTA格式的文件中,我们只需要打开该文件并将此记录作为字符串读取,并将其用作查询参数:
>>> from Bio import Blast
>>> fasta_string = open("m_cold.fasta").read()
>>> result_stream = Blast.qblast("blastn", "nt", fasta_string)
我们还可以在FASTA文件中读取为 SeqRecord 然后仅提供序列本身:
>>> from Bio import Blast
>>> from Bio import SeqIO
>>> record = SeqIO.read("m_cold.fasta", "fasta")
>>> result_stream = Blast.qblast("blastn", "nt", record.seq)
仅提供序列意味着BST将自动为您的序列分配标识符。您可能更愿意打电话 format 上 SeqRecord 创建FASTA字符串(其中将包括现有标识符)的对象:
>>> from Bio import Blast
>>> from Bio import SeqIO
>>> records = SeqIO.parse("ls_orchid.gbk", "genbank")
>>> record = next(records)
>>> result_stream = Blast.qblast("blastn", "nt", format(record, "fasta"))
如果您的序列采用非FASTA文件格式(您可以使用该格式提取),则这种方法更有意义 Bio.SeqIO (see章 序列输入/输出 ).
保存BST结果
无论你提出什么论点, qblast() 函数,您应该以流的形式返回结果 bytes 数据(默认情况下为ML格式)。下一步将是将HTML输出解析为表示搜索结果的Python对象(第节 解析BST输出 ),但您可能想先保存输出文件的本地副本。我发现这在调试从BST结果中提取信息的代码时特别有用(因为重新运行在线搜索速度慢并且浪费NCBI计算机时间)。
我们需要小心一点,因为我们可以使用 result_stream.read() 仅读取一次BST输出-调用 result_stream.read() 再次返回空的 bytes object.
>>> with open("my_blast.xml", "wb") as out_stream:
... out_stream.write(result_stream.read())
...
>>> result_stream.close()
完成此操作后,结果将保存在文件中 my_blast.xml 和 result_stream 已提取其所有数据(所以我们关闭了它)。但 parse AMPS解析器的功能(中描述 解析BST输出 )采用类似文件的对象,因此我们只需打开保存的文件以进行输入 bytes :
>>> result_stream = open("my_blast.xml", "rb")
既然我们已经将BST结果再次放回数据流中,我们就准备对它们做一些事情了,所以这将我们直接进入解析部分(请参阅部分 解析BST输出 下面)。您现在可能想直接跳到这一点……
获取其他格式的BST输出
通过使用 format_type 呼叫时的争论 qblast ,您可以以非HTML格式获取BST输出。以下是读取SON格式的AMPS输出的示例。使用 format_type="JSON2" ,由 Blast.qblast 将采用压缩的杨森格式:
>>> from Bio import Blast
>>> from Bio import SeqIO
>>> record = SeqIO.read("m_cold.fasta", "fasta")
>>> result_stream = Blast.qblast("blastn", "nt", record.seq, format_type="JSON2")
>>> data = result_stream.read()
>>> data[:4]
b'PK\x03\x04'
这是Zip文件的神奇数字。
>>> with open("myzipfile.zip", "wb") as out_stream:
... out_stream.write(data)
...
13813
请注意,我们将数据读写为 bytes .现在打开我们创建的Zip文件:
>>> import zipfile
>>> myzipfile = zipfile.ZipFile("myzipfile.zip")
>>> myzipfile.namelist()
['N5KN7UMJ013.json', 'N5KN7UMJ013_1.json']
>>> stream = myzipfile.open("N5KN7UMJ013.json")
>>> data = stream.read()
这些数据 bytes ,所以我们需要解码它们以获得字符串对象:
>>> data = data.decode()
>>> print(data)
{
"BlastJSON": [
{"File": "N5KN7UMJ013_1.json" }
]
}
现在打开Zip文件中包含的第二个文件,以获取杨森格式的BST结果:
>>> stream = myzipfile.open("N5KN7UMJ013_1.json")
>>> data = stream.read()
>>> len(data)
145707
>>> data = data.decode()
>>> print(data)
{
"BlastOutput2": {
"report": {
"program": "blastn",
"version": "BLASTN 2.14.1+",
"reference": "Stephen F. Altschul, Thomas L. Madden, Alejandro A. ...
"search_target": {
"db": "nt"
},
"params": {
"expect": 10,
"sc_match": 2,
"sc_mismatch": -3,
"gap_open": 5,
"gap_extend": 2,
"filter": "L;m;"
},
"results": {
"search": {
"query_id": "Query_69183",
"query_len": 1111,
"query_masking": [
{
"from": 797,
"to": 1110
}
],
"hits": [
{
"num": 1,
"description": [
{
"id": "gi|1219041180|ref|XM_021875076.1|",
...
我们可以使用Python标准库中的SON解析器将SON数据转换为常规Python字典:
>>> import json
>>> d = json.loads(data)
>>> print(d)
{'BlastOutput2': {'report': {'program': 'blastn', 'version': 'BLASTN 2.14.1+',
'reference': 'Stephen F. Altschul, Thomas L. Madden, Alejandro A. Schäffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped BLAST and PSI-BLAST: a new generation of protein database search programs",
Nucleic Acids Res. 25:3389-3402.',
'search_target': {'db': 'nt'}, 'params': {'expect': 10, 'sc_match': 2,
'sc_mismatch': -3, 'gap_open': 5, 'gap_extend': 2, 'filter': 'L;m;'},
'results': {'search': {'query_id': 'Query_128889', 'query_len': 1111,
'query_masking': [{'from': 797, 'to': 1110}], 'hits': [{'num': 1,
'description': [{'id': 'gi|1219041180|ref|XM_021875076.1|', 'accession':
'XM_021875076', 'title':
'PREDICTED: Chenopodium quinoa cold-regulated 413 plasma membrane protein 2-like (LOC110697660), mRNA',
'taxid': 63459, 'sciname': 'Chenopodium quinoa'}], 'len': 1173, 'hsps':
[{'num': 1, 'bit_score': 435.898, 'score': 482, 'evalue': 9.02832e-117,
'identity': 473, 'query_from'
...
本地运行AMPS
介绍
在本地运行BST(与通过互联网相反,请参阅第节 在互联网上运行AMPS )至少具有两个主要优势:
本地AMPS可能比互联网上的BST更快;
本地AMPS允许您创建自己的数据库来搜索序列。
处理专有或未发布的序列数据可能是本地运行RST的另一个原因。您可能不被允许重新分发序列,因此将它们作为BST查询提交给NCBI不是一种选择。
不幸的是,也有一些主要缺点-安装所有部分并正确设置需要一些努力:
本地AMPS需要安装命令行工具。
本地BLAST需要建立(大型)BLAST数据库(并可能保持最新)。
独立的NCBI AMPS +
“新” NCBI BLAST+ 套件于2009年发布。这将取代旧的NCBI“遗留”BST包(请参阅 其他版本的AMPS ).
本节将简要展示如何在Python中使用这些工具。如果您已经阅读或尝试过第部分中的对齐工具示例 对准工具 这一切看起来应该非常简单。首先,我们构建一个命令行字符串(就像手动运行独立的AMPS时您会在命令行提示符处输入的那样)。然后我们可以在Python内部执行此命令。
例如,获取基因核苷序列的FASTA文件,您可能想要针对非冗余(NR)蛋白质数据库运行BLASTX(翻译)搜索。假设您(或您的系统管理员)已下载并安装NR数据库,则您可能会运行:
$ blastx -query opuntia.fasta -db nr -out opuntia.xml -evalue 0.001 -outfmt 5
这应针对NR数据库运行BLASTX,使用预期截止值 \(0.001\) 并生成指定文件的ML输出(然后我们可以解析该文件)。在我的计算机上,这大约需要六分钟--这是将输出保存到文件的一个很好的理由,以便您可以根据需要重复任何分析。
在Python中,我们可以使用 subprocess 模块用于构建命令行字符串并运行它:
>>> import subprocess
>>> cmd = "blastx -query opuntia.fasta -db nr -out opuntia.xml"
>>> cmd += " -evalue 0.001 -outfmt 5"
>>> subprocess.run(cmd, shell=True)
在本例中,BLASTX不应该有任何输出从终端。您可能需要检查输出文件 opuntia.xml 已经创建。
您可能还记得本教程中的早期示例, opuntia.fasta 包含七个序列,因此BLASTML输出应该包含多个结果。因此使用 Bio.Blast.parse() 按以下部分中所述解析它 解析BST输出 .
其他版本的AMPS
NCBI BST+(用C++编写)于2009年首次发布,以替代不再更新的原始NCBI“遗留”BST(用C编写)。你可能也会遇到 Washington University BLAST (WU-BLAST)及其继任者, Advanced Biocomputing BLAST (AB-AMPS,2009年发布,非免费/开源)。这些包包括命令行工具 wu-blastall 和 ab-blastall ,它模仿了 blastall 来自NCBI“遗留”AMPS套件。Biopython目前没有提供用于调用这些工具的包装器,但应该能够解析它们的任何NCBI兼容输出。
解析BST输出
如上所述,BST可以生成各种格式的输出,例如HTML、HTML和纯文本。最初,Biopython拥有用于AMPS纯文本和HTML输出的解析器,因为这些是当时提供的唯一输出格式。这些解析器现已从Biopython中删除,因为这些格式的BST输出不断变化,每次都会破坏Biopython解析器。如今,Biopython可以解析HTML格式、ML2格式和表格格式的BST输出。本章描述使用 Bio.Blast.parse 功能该功能自动检测该文档是为ML格式还是为ML 2格式。表格格式的BST输出可以使用 Bio.Align.parse 功能(请参阅部分 来自AMPS或FASTA的表格输出 ).
您可以通过各种方式获得HTML格式的BST输出。对于解析器来说,输出是如何生成的并不重要,只要它是以HTML格式为即可。
您可以使用Biopython通过互联网运行BST,如部分所述 在互联网上运行AMPS .
您可以使用Biopython在本地运行BST,如部分所述 本地运行AMPS .
您可以通过网络浏览器在NCBI网站上进行AMPS搜索,然后保存结果。您需要选择ML作为接收结果的格式,并保存您获得的最终BST页面(您知道,包含所有有趣结果的页面!)到文件。
您还可以在不使用Biopython的情况下在本地运行BST,并将输出保存在文件中。同样,您需要选择ML作为接收结果的格式。
重要的一点是,您不必使用Biopython脚本来获取数据来解析它。以这些方式之一执行任务,您需要获得一个类似文件的对象来获取结果。在Python中,类似文件的对象或处理只是描述任何信息源的输入的一种很好的通用方式,以便可以使用使用检索信息 read() 和 readline() 功能(请参阅部分 把手到底是什么? ).
如果您遵循上面的代码通过脚本与AMPS交互,那么您已经拥有 result_stream ,将文件类对象转化为BST结果。例如,使用GI号进行在线搜索:
>>> from Bio import Blast
>>> result_stream = Blast.qblast("blastn", "nt", "8332116")
如果您以其他方式运行BST,并在文件中包含BST输出(以HTML格式) my_blast.xml ,您需要做的就是打开文件进行阅读(作为 bytes ):
>>> result_stream = open("my_blast.xml", "rb")
既然我们已经获得了数据流,我们就准备好解析输出了。解析它的代码确实相当小。如果您期望得到单个BST结果(即,您使用了一个查询):
>>> from Bio import Blast
>>> blast_record = Blast.read(result_stream)
或者,如果你有很多结果(即,多个查询序列):
>>> from Bio import Blast
>>> blast_records = Blast.parse(result_stream)
就像 Bio.SeqIO 和 Bio.Align (see章 序列输入/输出 和 序列比对 ),我们有一对输入函数, read 和 parse ,在哪里 read 当您只有一个对象时,并且 parse 是一个迭代器,用于当您可以拥有大量对象时-但不是获取 SeqRecord 或 Alignment 对象,我们就会得到AMPS记录对象。
为了能够处理AMPS文件可能很大、包含数千个结果的情况, Blast.parse() 返回迭代器。简而言之,迭代器允许您分步执行RST输出,为每个RST搜索结果逐一检索RST记录:
>>> from Bio import Blast
>>> blast_records = Blast.parse(result_stream)
>>> blast_record = next(blast_records)
# ... do something with blast_record
>>> blast_record = next(blast_records)
# ... do something with blast_record
>>> blast_record = next(blast_records)
# ... do something with blast_record
>>> blast_record = next(blast_records)
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
StopIteration
# No further records
或者,您可以使用 for -loop:
>>> for blast_record in blast_records:
... pass # Do something with blast_record
...
不过请注意,您只能浏览一次BST记录。通常,您会从每个RST记录中保存您感兴趣的信息。
也可以使用 blast_records 作为列表,例如通过索引提取一个记录,或者通过调用 len 或 print 对 blast_records .然后,解析器将自动重写记录并存储它们:
>>> from Bio import Blast
>>> blast_records = Blast.parse("xml_2222_blastx_001.xml")
>>> len(blast_records) # this causes the parser to iterate over all records
7
>>> blast_records[2].query.description
'gi|5690369|gb|AF158246.1|AF158246 Cricetulus griseus glucose phosphate isomerase (GPI) gene, partial intron sequence'
但如果您的AMPS文件很大,则在尝试将它们全部保存在列表中时可能会遇到内存问题。
如果您开始迭代记录 before 使用 blast_records 作为列表,解析器首先将文件流重置为数据的开头,以确保所有记录都需要读取。请注意,如果流无法重置为开头,例如如果正在远程读取数据(例如通过qblast;请参阅小节 在互联网上运行AMPS ).在这些情况下,您可以通过调用将记录显式地读取到列表中 blast_records = blast_records[:] 然后再迭代它们。在阅读记录后,可以安全地将其删除或将其用作列表。
您可以直接提供文件名,而不是自己打开文件:
>>> from Bio import Blast
>>> with Blast.parse("my_blast.xml") as blast_records:
... for blast_record in blast_records:
... pass # Do something with blast_record
...
在这种情况下,Biopython会为您打开文件,并在不再需要该文件时立即关闭它(虽然可以简单地使用 blast_records = Blast.parse("my_blast.xml") ,它的缺点是文件保持打开的时间可能比严格需要的时间更长,从而浪费资源)。
你可以 print 快速概述其内容的记录:
>>> from Bio import Blast
>>> with Blast.parse("my_blast.xml") as blast_records:
... print(blast_records)
...
Program: BLASTN 2.2.27+
db: refseq_rna
Query: 42291 (length=61)
mystery_seq
Hits: ---- ----- ----------------------------------------------------------
# # HSP ID + description
---- ----- ----------------------------------------------------------
0 1 gi|262205317|ref|NR_030195.1| Homo sapiens microRNA 52...
1 1 gi|301171311|ref|NR_035856.1| Pan troglodytes microRNA...
2 1 gi|270133242|ref|NR_032573.1| Macaca mulatta microRNA ...
3 2 gi|301171322|ref|NR_035857.1| Pan troglodytes microRNA...
4 1 gi|301171267|ref|NR_035851.1| Pan troglodytes microRNA...
5 2 gi|262205330|ref|NR_030198.1| Homo sapiens microRNA 52...
6 1 gi|262205302|ref|NR_030191.1| Homo sapiens microRNA 51...
7 1 gi|301171259|ref|NR_035850.1| Pan troglodytes microRNA...
8 1 gi|262205451|ref|NR_030222.1| Homo sapiens microRNA 51...
9 2 gi|301171447|ref|NR_035871.1| Pan troglodytes microRNA...
10 1 gi|301171276|ref|NR_035852.1| Pan troglodytes microRNA...
11 1 gi|262205290|ref|NR_030188.1| Homo sapiens microRNA 51...
12 1 gi|301171354|ref|NR_035860.1| Pan troglodytes microRNA...
13 1 gi|262205281|ref|NR_030186.1| Homo sapiens microRNA 52...
14 2 gi|262205298|ref|NR_030190.1| Homo sapiens microRNA 52...
15 1 gi|301171394|ref|NR_035865.1| Pan troglodytes microRNA...
16 1 gi|262205429|ref|NR_030218.1| Homo sapiens microRNA 51...
17 1 gi|262205423|ref|NR_030217.1| Homo sapiens microRNA 52...
18 1 gi|301171401|ref|NR_035866.1| Pan troglodytes microRNA...
19 1 gi|270133247|ref|NR_032574.1| Macaca mulatta microRNA ...
20 1 gi|262205309|ref|NR_030193.1| Homo sapiens microRNA 52...
21 2 gi|270132717|ref|NR_032716.1| Macaca mulatta microRNA ...
22 2 gi|301171437|ref|NR_035870.1| Pan troglodytes microRNA...
23 2 gi|270133306|ref|NR_032587.1| Macaca mulatta microRNA ...
24 2 gi|301171428|ref|NR_035869.1| Pan troglodytes microRNA...
25 1 gi|301171211|ref|NR_035845.1| Pan troglodytes microRNA...
26 2 gi|301171153|ref|NR_035838.1| Pan troglodytes microRNA...
27 2 gi|301171146|ref|NR_035837.1| Pan troglodytes microRNA...
28 2 gi|270133254|ref|NR_032575.1| Macaca mulatta microRNA ...
29 2 gi|262205445|ref|NR_030221.1| Homo sapiens microRNA 51...
~~~
97 1 gi|356517317|ref|XM_003527287.1| PREDICTED: Glycine ma...
98 1 gi|297814701|ref|XM_002875188.1| Arabidopsis lyrata su...
99 1 gi|397513516|ref|XM_003827011.1| PREDICTED: Pan panisc...
通常,您一次将运行一个BST搜索。然后,您需要做的就是拾取中的第一个(也是唯一一个)BST记录 blast_records :
>>> from Bio import Blast
>>> blast_records = Blast.parse("my_blast.xml")
>>> blast_record = next(blast_records)
或者更优雅地:
>>> from Bio import Blast
>>> blast_record = Blast.read(result_stream)
或者,等价地,
>>> from Bio import Blast
>>> blast_record = Blast.read("my_blast.xml")
(here,您不需要使用 with 块作为 Blast.read 将读取整个文件并随后立即关闭)。
我想现在你想知道BLAST记录中有什么。
BLAST Records、Record和Hit课程
BLAST Records课程
单个RST输出文件可以包含多个RST查询的输出。在Biopython中,AMPS输出文件中的信息存储在 Bio.Blast.Records object.这是一个返回一个的迭代器 Bio.Blast.Record 对象(参见小节 爆炸记录课程 )针对每个查询。的 Bio.Blast.Records 对象具有描述BLAST运行的以下属性:
source:用于Bio.Blast.Records已构建对象(这可以是文件名或路径,或者类似文件的对象)。program:所使用的特定BST程序(例如,“blastn”)。version:RST程序的版本(例如,' BLACK 2.2.27 +')。reference:对AMPS出版物的文献参考。db:运行查询所针对的BST数据库(例如,' nr ')。query:ASeqRecord对象可能包含以下部分或全部信息:query.id:查询的SeqId;query.description:查询的定义行;query.seq:查询序列。
param:包含用于BST运行的参数的字典。您可能会在本词典中找到以下关键:'matrix':BLAST运行中使用的评分矩阵(例如,“);'expect':预期机会匹配数量的阈值(浮动);'include':包含在psiblast(浮动)中多遍模型中的e值阈值;'sc-match':匹配核苷的分数(整);'sc-mismatch':不匹配的核苷的分数(整;'gap-open':缺口开盘成本(整);'gap-extend':缺口扩展成本(整);'filter':在BST运行中应用的过滤选项(字符串);'pattern':PHI-AMPS模式(字符串);'entrez-query':对Deliverz查询的请求限制(字符串)。
mbstat:一本包含Mega AMPS搜索统计数据的词典。请参阅Record.stat属性如下(小节中 爆炸记录课程 )以获取本词典中项目的描述。只有较旧版本的Mega BLAST存储此信息。由于该属性存储在靠近BST输出文件的结尾处,因此只能在文件被完全读取后(通过迭代记录直到StopIteration已发布)。
对于我们的例子,我们发现:
>>> blast_records
<Bio.Blast.Records source='my_blast.xml' program='blastn' version='BLASTN 2.2.27+' db='refseq_rna'>
>>> blast_records.source
'my_blast.xml'
>>> blast_records.program
'blastn'
>>> blast_records.version
'BLASTN 2.2.27+'
>>> blast_records.reference
'Stephen F. Altschul, Thomas L. Madden, Alejandro A. Schäffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res. 25:3389-3402.'
>>> blast_records.db
'refseq_rna'
>>> blast_records.param
{'expect': 10.0, 'sc-match': 2, 'sc-mismatch': -3, 'gap-open': 5, 'gap-extend': 2, 'filter': 'L;m;'}
>>> print(blast_records)
Program: BLASTN 2.2.27+
db: refseq_rna
Query: 42291 (length=61)
mystery_seq
Hits: ---- ----- ----------------------------------------------------------
# # HSP ID + description
---- ----- ----------------------------------------------------------
0 1 gi|262205317|ref|NR_030195.1| Homo sapiens microRNA 52...
1 1 gi|301171311|ref|NR_035856.1| Pan troglodytes microRNA...
2 1 gi|270133242|ref|NR_032573.1| Macaca mulatta microRNA ...
3 2 gi|301171322|ref|NR_035857.1| Pan troglodytes microRNA...
...
爆炸记录课程
A Bio.Blast.Record 对象存储由BST为单个查询提供的信息。的 Bio.Blast.Record 类继承自 list ,本质上是一个列表 Bio.Blast.Hit 对象(请参阅部分 BLAST Hit类 ).一 Bio.Blast.Record 对象具有以下两个属性:
query:ASeqRecord对象可能包含以下部分或全部信息:query.id:查询的SeqId;query.description:查询的定义行;query.seq:查询序列。
stat:一本包含AMPS命中统计数据的词典。您可能会在本词典中找到以下关键:'db-num':AMPS数据库中的序列数(integer);'db-len':AMPS数据库的长度(integer);'hsp-len':有效SPP(高分对)长度(整);'eff-space':有效搜索空间(浮动);'kappa':Karlin-Altschul参数K(浮动);'lambda':Karlin-Altschul参数Lambda(float);'entropy':Karlin-Altschul参数H(浮点)
message:一些(错误?)信息.
继续我们的例子,
>>> blast_record
<Bio.Blast.Record query.id='42291'; 100 hits>
>>> blast_record.query
SeqRecord(seq=Seq(None, length=61), id='42291', name='<unknown name>', description='mystery_seq', dbxrefs=[])
>>> blast_record.stat
{'db-num': 3056429, 'db-len': 673143725, 'hsp-len': 0, 'eff-space': 0, 'kappa': 0.41, 'lambda': 0.625, 'entropy': 0.78}
>>> print(blast_record)
Query: 42291 (length=61)
mystery_seq
Hits: ---- ----- ----------------------------------------------------------
# # HSP ID + description
---- ----- ----------------------------------------------------------
0 1 gi|262205317|ref|NR_030195.1| Homo sapiens microRNA 52...
1 1 gi|301171311|ref|NR_035856.1| Pan troglodytes microRNA...
2 1 gi|270133242|ref|NR_032573.1| Macaca mulatta microRNA ...
3 2 gi|301171322|ref|NR_035857.1| Pan troglodytes microRNA...
...
为 Bio.Blast.Record 类继承自 list ,您可以这样使用它。例如,您可以重写记录:
>>> for hit in blast_record:
... hit
...
<Bio.Blast.Hit target.id='gi|262205317|ref|NR_030195.1|' query.id='42291'; 1 HSP>
<Bio.Blast.Hit target.id='gi|301171311|ref|NR_035856.1|' query.id='42291'; 1 HSP>
<Bio.Blast.Hit target.id='gi|270133242|ref|NR_032573.1|' query.id='42291'; 1 HSP>
<Bio.Blast.Hit target.id='gi|301171322|ref|NR_035857.1|' query.id='42291'; 2 HSPs>
<Bio.Blast.Hit target.id='gi|301171267|ref|NR_035851.1|' query.id='42291'; 1 HSP>
...
要检查有多少次点击 blast_record 有,您可以简单地调用Python的 len 功能:
>>> len(blast_record)
100
与Python列表一样,您可以从 Bio.Blast.Record 使用指数:
>>> blast_record[0] # retrieves the top hit
<Bio.Blast.Hit target.id='gi|262205317|ref|NR_030195.1|' query.id='42291'; 1 HSP>
>>> blast_record[-1] # retrieves the last hit
<Bio.Blast.Hit target.id='gi|397513516|ref|XM_003827011.1|' query.id='42291'; 1 HSP>
检索多个匹配项 Bio.Blast.Record ,您可以使用切片符号。这将返回新的 Bio.Blast.Record 只包含切片命中的对象:
>>> blast_slice = blast_record[:3] # slices the first three hits
>>> print(blast_slice)
Query: 42291 (length=61)
mystery_seq
Hits: ---- ----- ----------------------------------------------------------
# # HSP ID + description
---- ----- ----------------------------------------------------------
0 1 gi|262205317|ref|NR_030195.1| Homo sapiens microRNA 52...
1 1 gi|301171311|ref|NR_035856.1| Pan troglodytes microRNA...
2 1 gi|270133242|ref|NR_032573.1| Macaca mulatta microRNA ...
要创建 Bio.Blast.Record ,取完整的切片:
>>> blast_record_copy = blast_record[:]
>>> type(blast_record_copy)
<class 'Bio.Blast.Record'>
>>> blast_record_copy # list of all hits
<Bio.Blast.Record query.id='42291'; 100 hits>
如果您想要对AMPS记录进行排序或过滤,这尤其有用(请参阅 排序和过滤BST输出 ),但希望保留原始AMPS输出的副本。
您还可以访问 blast_record 作为Python字典,并使用命中的ID作为键检索命中:
>>> blast_record["gi|262205317|ref|NR_030195.1|"]
<Bio.Blast.Hit target.id='gi|262205317|ref|NR_030195.1|' query.id='42291'; 1 HSP>
如果在 blast_record 、a KeyError 提出:
>>> blast_record["unicorn_gene"]
Traceback (most recent call last):
...
KeyError: 'unicorn_gene'
您可以通过使用 .keys() 像往常一样:
>>> blast_record.keys()
['gi|262205317|ref|NR_030195.1|', 'gi|301171311|ref|NR_035856.1|', 'gi|270133242|ref|NR_032573.1|', ...]
如果您只是想检查查询结果中是否存在特定的命中,该怎么办?您可以使用进行简单的Python成员资格测试 in 关键字:
>>> "gi|262205317|ref|NR_030195.1|" in blast_record
True
>>> "gi|262205317|ref|NR_030194.1|" in blast_record
False
有时,仅仅知道热门歌曲是否存在是不够的;您还想知道热门歌曲的排名。这里 index 方法来拯救:
>>> blast_record.index("gi|301171437|ref|NR_035870.1|")
22
请记住,Python使用从零开始的索引,因此第一次命中将位于索引0处。
BLAST Hit类
每个 Bio.Blast.Hit 中对象 blast_record 列表表示查询对目标的一次BST命中。
>>> hit = blast_record[0]
>>> hit
<Bio.Blast.Hit target.id='gi|262205317|ref|NR_030195.1|' query.id='42291'; 1 HSP>
>>> hit.target
SeqRecord(seq=Seq(None, length=61), id='gi|262205317|ref|NR_030195.1|', name='NR_030195', description='Homo sapiens microRNA 520b (MIR520B), microRNA', dbxrefs=[])
我们可以得到的摘要 hit 通过打印:
>>> print(blast_record[3])
Query: 42291
mystery_seq
Hit: gi|301171322|ref|NR_035857.1| (length=86)
Pan troglodytes microRNA mir-520c (MIR520C), microRNA
HSPs: ---- -------- --------- ------ --------------- ---------------------
# E-value Bit score Span Query range Hit range
---- -------- --------- ------ --------------- ---------------------
0 8.9e-20 100.47 60 [1:61] [13:73]
1 3.3e-06 55.39 60 [0:60] [73:13]
您可以看到我们在这里涵盖了必需品:
命中始终针对一个查询;查询ID和描述显示在摘要的顶部。
命中由查询与一个目标序列的一个或多个比对组成。接下来显示的目标信息是摘要。如上所示,可以通过
target命中的属性。最后,有一个表格,包含有关每次点击包含的对齐的快速信息。用AMPS的说法,这些比对被称为“高分片段对”或HSPs(请参阅部分 BLAST热休克蛋白类 ).表中的每一行总结了一个热稳定点,包括热稳定点指数、e值、位得分、跨度(包括间隙的对齐长度)、查询坐标和目标坐标。
的 Bio.Blast.Hit 类是 Bio.Align.Alignments (复数;请参阅部分 Alignments课程 ),因此本质上是一个列表 Bio.Align.Alignment (单数;见部分 路线对象 )对象。特别是当将核苷序列与基因组进行比对时, Bio.Blast.Hit 对象可能由多个组成 Bio.Align.Alignment 如果特定查询与染色体的多个区域对齐。对于蛋白质比对,通常一次命中仅由一次比对组成,尤其是对于高度相似序列的比对。
>>> type(hit)
<class 'Bio.Blast.Hit'>
>>> from Bio.Align import Alignments
>>> isinstance(hit, Alignments)
True
>>> len(hit)
1
对于ML 2格式的BST输出,命中可能具有多个序列相同但序列ID和描述不同的目标。这些目标可以作为 hit.targets 属性在大多数情况下, hit.targets 长度为1且仅包含 hit.target :
>>> from Bio import Blast
>>> blast_record = Blast.read("xml_2900_blastx_001_v2.xml")
>>> for hit in blast_record:
... print(len(hit.targets))
...
1
1
2
1
1
1
1
1
1
1
然而,正如您在上面的输出中看到的那样,第三次命中有多个目标。
>>> hit = blast_record[2]
>>> hit.targets[0].seq
Seq(None, length=246)
>>> hit.targets[1].seq
Seq(None, length=246)
>>> hit.targets[0].id
'gi|684409690|ref|XP_009175831.1|'
>>> hit.targets[1].id
'gi|663044098|gb|KER20427.1|'
>>> hit.targets[0].name
'XP_009175831'
>>> hit.targets[1].name
'KER20427'
>>> hit.targets[0].description
'hypothetical protein T265_11027 [Opisthorchis viverrini]'
>>> hit.targets[1].description
'hypothetical protein T265_11027 [Opisthorchis viverrini]'
由于两个目标的序列内容彼此相同,因此它们的序列比对也相同。因此,这次点击的对齐仅指 hit.targets[0] (与 hit.target ),作为对齐 hit.targets[1] 无论如何都会是一样的。
BLAST热休克蛋白类
让我们回到我们的主要示例,看看第一次命中中的第一个(也是唯一的)对齐。此对齐方式是 Bio.Blast.HSP 类,它是 Alignment 阶级 Bio.Align :
>>> from Bio import Blast
>>> blast_record = Blast.read("my_blast.xml")
>>> hit = blast_record[0]
>>> len(hit)
1
>>> alignment = hit[0]
>>> alignment
<Bio.Blast.HSP target.id='gi|262205317|ref|NR_030195.1|' query.id='42291'; 2 rows x 61 columns>
>>> type(alignment)
<class 'Bio.Blast.HSP'>
>>> from Bio.Align import Alignment
>>> isinstance(alignment, Alignment)
True
的 alignment 对象具有指向目标和查询序列的属性,以及 coordinates 描述序列比对的属性。
>>> alignment.target
SeqRecord(seq=Seq('CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCCTTT...GGG'), id='gi|262205317|ref|NR_030195.1|', name='NR_030195', description='Homo sapiens microRNA 520b (MIR520B), microRNA', dbxrefs=[])
>>> alignment.query
SeqRecord(seq=Seq('CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCCTTT...GGG'), id='42291', name='<unknown name>', description='mystery_seq', dbxrefs=[])
>>> alignment.target
SeqRecord(seq=Seq('CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCCTTT...GGG'), id='gi|262205317|ref|NR_030195.1|', name='NR_030195', description='Homo sapiens microRNA 520b (MIR520B), microRNA', dbxrefs=[])
>>> alignment.query
SeqRecord(seq=Seq('CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCCTTT...GGG'), id='42291', name='<unknown name>', description='mystery_seq', dbxrefs=[])
>>> print(alignment.coordinates)
[[ 0 61]
[ 0 61]]
对于翻译的BST搜索, features 目标或查询的属性可能包含 SeqFeature 存储氨基酸序列区域的CDS型。的 qualifiers 此类特征的属性是具有单字的字典 'coded_by' ;相应的值指定编码氨基酸序列的核苷酸序列区域,位于以1为基础的坐标的班克风格字符串中。
每个 Alignment 对象具有以下附加属性:
score:高分对(SPP)的分数;annotations:可能包含以下键的字典:'bit score':SPP(浮动)的分数(以位为单位);'evalue':SPP的e值(浮动);'identity':SPP中的身份数(integer);'positive':热休克蛋白阳性数(整);'gaps':热休克蛋白中的间隙数(整);'midline':格式化中间行。
通常的 Alignment 方法(请参阅部分 路线对象 )可以应用于 alignment .例如,我们可以打印对齐方式:
>>> print(alignment)
Query : 42291 Length: 61 Strand: Plus
mystery_seq
Target: gi|262205317|ref|NR_030195.1| Length: 61 Strand: Plus
Homo sapiens microRNA 520b (MIR520B), microRNA
Score:111 bits(122), Expect:5e-23,
Identities:61/61(100%), Positives:61/61(100%), Gaps:0.61(0%)
gi|262205 0 CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCCTTTTAGAGG
0 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
42291 0 CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCCTTTTAGAGG
gi|262205 60 G 61
60 | 61
42291 60 G 61
让我们打印出有关我们的AMPS记录中所有大于特定阈值的点击的一些摘要信息:
>>> E_VALUE_THRESH = 0.04
>>> for alignments in blast_record:
... for alignment in alignments:
... if alignment.annotations["evalue"] < E_VALUE_THRESH:
... print("****Alignment****")
... print("sequence:", alignment.target.id, alignment.target.description)
... print("length:", len(alignment.target))
... print("score:", alignment.score)
... print("e value:", alignment.annotations["evalue"])
... print(alignment[:, :50])
...
****Alignment****
sequence: gi|262205317|ref|NR_030195.1| Homo sapiens microRNA 520b (MIR520B), microRNA
length: 61
score: 122.0
e value: 4.91307e-23
gi|262205 0 CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTC 50
0 |||||||||||||||||||||||||||||||||||||||||||||||||| 50
42291 0 CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTC 50
****Alignment****
sequence: gi|301171311|ref|NR_035856.1| Pan troglodytes microRNA mir-520b (MIR520B), microRNA
length: 60
score: 120.0
e value: 1.71483e-22
gi|301171 0 CCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCC 50
0 |||||||||||||||||||||||||||||||||||||||||||||||||| 50
42291 1 CCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTCC 51
****Alignment****
sequence: gi|270133242|ref|NR_032573.1| Macaca mulatta microRNA mir-519a (MIR519A), microRNA
length: 85
score: 112.0
e value: 2.54503e-20
gi|270133 12 CCCTCTAGAGGGAAGCGCTTTCTGTGGTCTGAAAGAAAAGAAAGTGCTTC 62
0 |||||||.|||||||||||||||||.|||||||||||||||||||||||| 50
42291 0 CCCTCTACAGGGAAGCGCTTTCTGTTGTCTGAAAGAAAAGAAAGTGCTTC 50
...
排序和过滤BST输出
如果RST输出文件中命中的顺序不适合您的口味,则可以使用 sort 在 Bio.Blast.Record object.例如,这里我们根据每个目标的序列长度对命中进行排序,并设置 reverse 标志以 True 以便我们按降序排序。
>>> for hit in blast_record[:5]:
... print(f"{hit.target.id} {len(hit.target)}")
...
gi|262205317|ref|NR_030195.1| 61
gi|301171311|ref|NR_035856.1| 60
gi|270133242|ref|NR_032573.1| 85
gi|301171322|ref|NR_035857.1| 86
gi|301171267|ref|NR_035851.1| 80
>>> sort_key = lambda hit: len(hit.target)
>>> blast_record.sort(key=sort_key, reverse=True)
>>> for hit in blast_recordt[:5]:
... print(f"{hit.target.id} {len(hit.target)}")
...
gi|397513516|ref|XM_003827011.1| 6002
gi|390332045|ref|XM_776818.2| 4082
gi|390332043|ref|XM_003723358.1| 4079
gi|356517317|ref|XM_003527287.1| 3251
gi|356543101|ref|XM_003539954.1| 2936
这会排序 blast_record 到位使用 original_blast_record = blast_record[:] 如果您想保留原始未排序的BST输出的副本,请在排序之前进行。
要根据属性过滤BST命中,您可以使用Python的内置 filter 使用适当的回调函数来评估每个命中。回调函数必须接受单个 Hit 对象并返回 True 或 False . 这是一个我们过滤掉的例子 Hit 只有一个热休克蛋白的对象:
>>> filter_func = lambda hit: len(hit) > 1 # the callback function
>>> len(blast_record) # no. of hits before filtering
100
>>> blast_record[:] = filter(filter_func, blast_record)
>>> len(blast_record) # no. of hits after filtering
37
>>> for hit in blast_record[:5]: # quick check for the hit lengths
... print(f"{hit.target.id} {len(hit)}")
...
gi|301171322|ref|NR_035857.1| 2
gi|262205330|ref|NR_030198.1| 2
gi|301171447|ref|NR_035871.1| 2
gi|262205298|ref|NR_030190.1| 2
gi|270132717|ref|NR_032716.1| 2
同样,您可以过滤每次点击中的热休克蛋白,例如根据其电子值:
>>> filter_func = lambda hsp: hsp.annotations["evalue"] < 1.0e-12
>>> for hit in blast_record:
... hit[:] = filter(filter_func, hit)
...
可能您想通过删除所有未剩余热休克蛋白的点击来跟进此操作:
>>> filter_func = lambda hit: len(hit) > 0
>>> blast_record[:] = filter(filter_func, blast_record)
>>> len(blast_record)
16
使用Python的内置 map 用于修改AMPS记录中的命中或热休克蛋白的功能。的 map 函数接受返回修改后的命中对象的回调函数。例如,我们可以使用 map 要重命名热门ID:
>>> for hit in blast_record[:5]:
... print(hit.target.id)
...
gi|301171322|ref|NR_035857.1|
gi|262205330|ref|NR_030198.1|
gi|301171447|ref|NR_035871.1|
gi|262205298|ref|NR_030190.1|
gi|270132717|ref|NR_032716.1|
>>> import copy
>>> original_blast_record = copy.deepcopy(blast_record)
>>> def map_func(hit):
... # renames "gi|301171322|ref|NR_035857.1|" to "NR_035857.1"
... hit.target.id = hit.target.id.split("|")[3]
... return hit
...
>>> blast_record[:] = map(map_func, blast_record)
>>> for hit in blast_record[:5]:
... print(hit.target.id)
...
NR_035857.1
NR_030198.1
NR_035871.1
NR_030190.1
NR_032716.1
>>> for hit in original_blast_record[:5]:
... print(hit.target.id)
...
gi|301171322|ref|NR_035857.1|
gi|262205330|ref|NR_030198.1|
gi|301171447|ref|NR_035871.1|
gi|262205298|ref|NR_030190.1|
gi|270132717|ref|NR_032716.1|
请注意,在这个例子中, map_func 就地修改命中。与排序和过滤(见上文)相反,使用 original_blast_record = blast_record[:] 不足以保留未修改的BLAST记录的副本,因为它创建了BLAST记录的浅副本,由指向相同的指针组成 Hit 对象相反,我们使用 copy.deepcopy 创建一个AMPS记录的副本,其中每个 Hit 对象被复制。
编写BLAST记录
使用 write 功能 Bio.Blast 将RST记录保存为ML文件。默认情况下,使用(基于DTS)的ML格式;您还可以使用 fmt="XML2" 论点 write 功能
>>> from Bio import Blast
>>> stream = Blast.qblast("blastn", "nt", "8332116")
>>> records = Blast.parse(stream)
>>> Blast.write(records, "my_qblast_output.xml")
or
>>> Blast.write(records, "my_qblast_output.xml", fmt="XML2")
在本示例中,我们可以保存由 Blast.qblast 直接到一个HTML文件(请参阅部分 保存BST结果 ).然而,通过将qblast返回的数据解析为记录,我们可以在保存OPS记录之前对它们进行排序或过滤。例如,我们可能只对积极评分至少为400的AMPS感兴趣:
>>> filter_func = lambda hsp: hsp.annotations["positive"] >= 400
>>> for hit in records[0]:
... hit[:] = filter(filter_func, hit)
...
>>> Blast.write(records, "my_qblast_output_selected.xml")
不是文件名,而是第二个参数 Blast.write 也可以是文件流。在这种情况下,必须以二进制格式打开流才能写入:
>>> with open("my_qblast_output.xml", "wb") as stream:
... Blast.write(records, stream)
...
处理PSI-AMPS
您可以运行PSI-AMPS的独立版本 (psiblast )直接从命令行或使用Python的 subprocess module.
在撰写本文时,NCBI似乎不支持通过互联网运行PSI-AMPS搜索的工具。
注意到 Bio.Blast 解析器可以从当前版本的PSI-BST读取ML输出,但诸如每次迭代中哪些序列是新的或重复使用的信息并不存在于该文件中。
处理RPS-AMPS
您可以运行RPS-AMPS的独立版本 (rpsblast )直接从命令行或使用Python的 subprocess module.
在撰写本文时,NCBI似乎不支持通过互联网运行RPS-BLAST搜索的工具。
您可以使用 Bio.Blast 解析器用于从当前版本的RPS-BST读取ML输出。
BLAST(旧)
嘿,每个人都喜欢BRAST吧?我的意思是,天哪,如何更容易地在你的一个序列与已知世界中的所有其他序列之间进行比较呢?但是,当然,这一部分并不是关于AMPS有多酷,因为我们已经知道这一点。这是关于BST的问题--处理大规模运行产生的大量数据以及一般来说自动化BST运行可能确实很困难。
幸运的是,Biopython的人们非常了解这一点,所以他们开发了很多工具来处理AMPS并让事情变得更容易。本节详细介绍了如何使用这些工具并使用它们做有用的事情。
处理AMPS可以分为两个步骤,这两个步骤都可以在Biopython内完成。首先,对您的查询序列运行BST,并获得一些输出。其次,在Python中解析BST输出以进行进一步分析。
您第一次了解运行AMPS可能是通过NCBI网络服务。事实上,有很多种方法可以运行AMPS,这些方法可以分为多种方式。最重要的区别是在本地运行BST(在您自己的机器上)和远程运行BST(在另一台机器上,通常是NCBI服务器)。我们将通过从Python脚本中调用NCBI在线BST服务来开始本章。
NOTE :以下章节 AMPS和其他序列搜索工具 描述 Bio.SearchIO .我们打算最终取代旧的 Bio.Blast 模块,因为它提供了一个更通用的框架,处理其他相关的序列搜索工具。但是,现在您可以使用它或旧的 Bio.Blast 用于处理NCBI AMPS的模块。
在互联网上运行AMPS
我们使用该功能 qblast() 在 Bio.Blast.NCBIWWW 调用在线版本的AMPS模块。这有三个非可选参数:
第一个参数是用于搜索的blast程序,作为大写字符串。程序的选项和描述可在https://blast.ncbi.nlm.nih.gov/Blast.cgi上获取。目前
qblast仅适用于blastup、blastp、blastx、tblast和tblastx。第二个参数指定要搜索的数据库。同样,该选项可在NCBI BST指南https://blast.ncbi.nlm.nih.gov/doc/blast-help/上找到。
第三个参数是包含查询序列的字符串。这可以是序列本身、fasta格式的序列,也可以是GI号等标识符。
来自https://blast.ncbi.nlm.nih.gov/doc/blast-help/developerinfo.html#developerinfo的NCBI指南规定:
联系服务器的频率不要超过每10秒一次。
对任何单个DID进行民意调查的频率不要超过每分钟一次。
使用URL参数电子邮件和工具,以便NCBI可以在出现问题时与您联系。
如果提交超过50个搜索,则在周末或工作日东部时间晚上9点至凌晨5点之间运行脚本。
为了满足第三点,可以设置 NCBIWWW.email 变量
>>> from Bio.Blast import NCBIWWW
>>> NCBIWWW.email = "A.N.Other@example.com"
的 qblast 函数还接受许多其他选项参数,这些参数基本上类似于您可以在AMPS网页上设置的不同参数。我们将在这里重点介绍其中一些:
的论点
url_base设置在互联网上运行BST的基本URL。默认情况下,它连接到NCBI。的
qblast函数可以以各种格式返回BST结果,您可以通过可选的format_type关键字:"HTML","Text","ASN.1",或者"XML".默认值为"XML",因为这是解析器期望的格式,如部分所述 解析BST输出 下面的论点
expect设置期望或e值阈值。
有关可选的BST参数的更多信息,请参阅NCBI自己的文档或Biopython中内置的文档:
>>> from Bio.Blast import NCBIWWW
>>> help(NCBIWWW.qblast)
请注意,NCBI BST网站上的默认设置与QBST上的默认设置不完全相同。如果得到不同的结果,您需要检查参数(例如,期望值阈值和差距值)。
例如,如果您有一个核苷酸序列,您想使用BLACKS在核苷酸数据库(nt)中搜索,并且您知道查询序列的GI编号,则可以用途:
>>> from Bio.Blast import NCBIWWW
>>> result_handle = NCBIWWW.qblast("blastn", "nt", "8332116")
或者,如果我们的查询序列已经在FASTA格式的文件中,我们只需要打开该文件并将此记录作为字符串读取,并将其用作查询参数:
>>> from Bio.Blast import NCBIWWW
>>> fasta_string = open("m_cold.fasta").read()
>>> result_handle = NCBIWWW.qblast("blastn", "nt", fasta_string)
我们还可以在FASTA文件中读取为 SeqRecord 然后仅提供序列本身:
>>> from Bio.Blast import NCBIWWW
>>> from Bio import SeqIO
>>> record = SeqIO.read("m_cold.fasta", format="fasta")
>>> result_handle = NCBIWWW.qblast("blastn", "nt", record.seq)
仅提供序列意味着BST将自动为您的序列分配标识符。您可能更喜欢使用 SeqRecord 创建FASTA字符串(其中将包括现有标识符)的对象格式方法:
>>> from Bio.Blast import NCBIWWW
>>> from Bio import SeqIO
>>> record = SeqIO.read("m_cold.fasta", format="fasta")
>>> result_handle = NCBIWWW.qblast("blastn", "nt", record.format("fasta"))
如果您的序列采用非FASTA文件格式(您可以使用该格式提取),则这种方法更有意义 Bio.SeqIO (see章 序列输入/输出 ).
无论你提出什么论点, qblast() 函数中,您应该在处理对象中返回结果(默认情况下为ML格式)。下一步将是将HTML输出解析为表示搜索结果的Python对象(第节 解析BST输出 ),但您可能想先保存输出文件的本地副本。我发现这在调试从BST结果中提取信息的代码时特别有用(因为重新运行在线搜索速度慢并且浪费NCBI计算机时间)。
我们需要小心一点,因为我们可以使用 result_handle.read() 仅读取一次BST输出-调用 result_handle.read() 再次返回空字符串。
>>> with open("my_blast.xml", "w") as out_handle:
... out_handle.write(result_handle.read())
...
>>> result_handle.close()
完成此操作后,结果将保存在文件中 my_blast.xml 并且原始手柄的所有数据已被提取(所以我们关闭了它)。但 parse AMPS解析器的功能(中描述 解析BST输出 )接受一个类似文件手柄的对象,因此我们只需打开保存的文件进行输入:
>>> result_handle = open("my_blast.xml")
既然我们已经将BST结果再次放回处理中,我们就准备好对它们执行一些操作了,所以这将我们直接进入解析部分(请参阅部分 解析BST输出 下面)。您现在可能想直接跳到这一点……
本地运行AMPS
介绍
在本地运行BST(与通过互联网相反,请参阅第节 在互联网上运行AMPS )至少具有两个主要优势:
本地AMPS可能比互联网上的BST更快;
本地AMPS允许您创建自己的数据库来搜索序列。
处理专有或未发布的序列数据可能是本地运行RST的另一个原因。您可能不被允许重新分发序列,因此将它们作为BST查询提交给NCBI不是一种选择。
不幸的是,也有一些主要缺点-安装所有部分并正确设置需要一些努力:
本地AMPS需要安装命令行工具。
本地BLAST需要建立(大型)BLAST数据库(并可能保持最新)。
为了进一步混淆问题,有几种不同的BST包可用,而且还有其他工具可以产生模仿的BST输出文件,例如BLAT。
独立的NCBI AMPS +
“新” NCBI BLAST+ 套件于2009年发布。这取代了旧的NCBI“遗留”AMPS包(见下文)。
本节将简要展示如何在Python中使用这些工具。如果您已经阅读或尝试过第部分中的对齐工具示例 对准工具 这一切看起来应该非常简单。首先,我们构建一个命令行字符串(就像手动运行独立的AMPS时您会在命令行提示符处输入的那样)。然后我们可以在Python内部执行此命令。
例如,获取基因核苷序列的FASTA文件,您可能想要针对非冗余(NR)蛋白质数据库运行BLASTX(翻译)搜索。假设您(或您的系统管理员)已下载并安装NR数据库,则您可能会运行:
$ blastx -query opuntia.fasta -db nr -out opuntia.xml -evalue 0.001 -outfmt 5
这应针对NR数据库运行BLASTX,使用预期截止值 \(0.001\) 并生成指定文件的ML输出(然后我们可以解析该文件)。在我的计算机上,这大约需要六分钟--这是将输出保存到文件的一个很好的理由,以便您可以根据需要重复任何分析。
在Python中,我们可以使用 subprocess 模块用于构建命令行字符串并运行它:
>>> import subprocess
>>> cmd = "blastx -query opuntia.fasta -db nr -out opuntia.xml"
>>> cmd += " -evalue 0.001 -outfmt 5"
>>> subprocess.run(cmd, shell=True)
在本例中,BLASTX不应该有任何输出从终端。您可能需要检查输出文件 opuntia.xml 已经创建。
您可能还记得本教程中的早期示例, opuntia.fasta 包含七个序列,因此BLASTML输出应该包含多个结果。因此使用 Bio.Blast.NCBIXML.parse() 按以下部分中所述解析它 解析BST输出 .
其他版本的AMPS
NCBI BST+(用C++编写)于2009年首次发布,以替代不再更新的原始NCBI“遗留”BST(用C编写)。有很多变化-旧版本只有一个核心命令行工具 blastall 它涵盖了多种不同的BST搜索类型(现在在RST+中是单独的命令),并且所有命令行选项都已重新命名。Biopython针对NCBI“遗留”BST工具的包装器已被废弃,并将在未来的版本中删除。为了避免混淆,我们在本教程中不涉及从Biopython调用这些旧工具。
你可能也会遇到 Washington University BLAST (WU-BLAST)及其继任者, Advanced Biocomputing BLAST (AB-AMPS,2009年发布,非免费/开源)。这些包包括命令行工具 wu-blastall 和 ab-blastall ,它模仿了 blastall 来自NCBI“遗留”AMPS套件。Biopython目前没有提供用于调用这些工具的包装器,但应该能够解析它们的任何NCBI兼容输出。
解析BST输出
如上所述,BST可以生成各种格式的输出,例如HTML、HTML和纯文本。最初,Biopython拥有用于AMPS纯文本和HTML输出的解析器,因为这些是当时提供的唯一输出格式。不幸的是,这些格式的BST输出不断变化,每次都会破坏Biopython解析器。我们的HTMLBLAST解析器已被删除,而废弃的纯文本BLAST解析器现在只能通过 Bio.SearchIO .使用它的风险由您自行承担,它可能会起作用,也可能不会起作用,具体取决于您使用的是哪一个BST版本。
由于跟上AMPS的变化已成为一项无望的努力,特别是对于运行不同的BST版本的用户,我们现在建议解析可由最新版本的BST生成的可扩展扩展格式的输出。不仅,ML输出比纯文本和HTML输出更稳定,而且自动解析也更容易,使Biopython更加稳定。
您可以通过各种方式获得HTML格式的BST输出。对于解析器来说,输出是如何生成的并不重要,只要它是以HTML格式为即可。
您可以使用Biopython通过互联网运行BST,如部分所述 在互联网上运行AMPS .
您可以使用Biopython在本地运行BST,如部分所述 本地运行AMPS .
您可以通过网络浏览器在NCBI网站上进行AMPS搜索,然后保存结果。您需要选择ML作为接收结果的格式,并保存您获得的最终BST页面(您知道,包含所有有趣结果的页面!)到文件。
您还可以在不使用Biopython的情况下在本地运行BST,并将输出保存在文件中。同样,您需要选择ML作为接收结果的格式。
重要的一点是,您不必使用Biopython脚本来获取数据来解析它。以这些方式之一执行任务,您需要处理结果。在Python中,手柄只是描述任何信息源的输入的一种很好的通用方式,以便可以使用使用检索信息 read() 和 readline() 功能(请参阅部分 把手到底是什么? ).
如果您遵循上面的代码通过脚本与AMPS交互,那么您已经拥有 result_handle ,BST结果的处理。例如,使用GI号进行在线搜索:
>>> from Bio.Blast import NCBIWWW
>>> result_handle = NCBIWWW.qblast("blastn", "nt", "8332116")
如果您以其他方式运行BST,并在文件中包含BST输出(以HTML格式) my_blast.xml ,您需要做的就是打开文件进行阅读:
>>> result_handle = open("my_blast.xml")
现在我们已经有了一个手柄,我们就准备好解析输出了。解析它的代码确实相当小。如果您期望得到单个BST结果(即,您使用了一个查询):
>>> from Bio.Blast import NCBIXML
>>> blast_record = NCBIXML.read(result_handle)
或者,如果你有很多结果(即,多个查询序列):
>>> from Bio.Blast import NCBIXML
>>> blast_records = NCBIXML.parse(result_handle)
就像 Bio.SeqIO 和 Bio.Align (see章 序列输入/输出 和 序列比对 ),我们有一对输入函数, read 和 parse ,在哪里 read 当您只有一个对象时,并且 parse 是一个迭代器,用于当您可以拥有大量对象时-但不是获取 SeqRecord 或 MultipleSeqAlignment 对象,我们就会得到AMPS记录对象。
为了能够处理AMPS文件可能很大、包含数千个结果的情况, NCBIXML.parse() 返回迭代器。简而言之,迭代器允许您分步执行RST输出,为每个RST搜索结果逐一检索RST记录:
>>> from Bio.Blast import NCBIXML
>>> blast_records = NCBIXML.parse(result_handle)
>>> blast_record = next(blast_records)
# ... do something with blast_record
>>> blast_record = next(blast_records)
# ... do something with blast_record
>>> blast_record = next(blast_records)
# ... do something with blast_record
>>> blast_record = next(blast_records)
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
StopIteration
# No further records
或者,您可以使用 for -loop:
>>> for blast_record in blast_records:
... pass # Do something with blast_record
...
不过请注意,您只能浏览一次BST记录。通常,您会从每个RST记录中保存您感兴趣的信息。如果您想保存所有返回的BST记录,可以将迭代器转换为列表:
>>> blast_records = list(blast_records)
现在您可以像往常一样使用索引访问列表中的每个BLAST记录。但如果您的AMPS文件很大,则在尝试将它们全部保存在列表中时可能会遇到内存问题。
通常,您一次将运行一个BST搜索。然后,您需要做的就是拾取中的第一个(也是唯一一个)BST记录 blast_records :
>>> from Bio.Blast import NCBIXML
>>> blast_records = NCBIXML.parse(result_handle)
>>> blast_record = next(blast_records)
或者更优雅地:
>>> from Bio.Blast import NCBIXML
>>> blast_record = NCBIXML.read(result_handle)
我想现在你想知道BLAST记录中有什么。
BLAST唱片班
AMPS记录包含您可能想要从AMPS输出中提取的所有内容。现在我们只展示如何从AMPS报告中获取一些信息的示例,但如果您想要这里没有描述的特定信息,请详细查看记录类上的信息,并仔细研究代码或自动生成的文档-文档字符串有很多关于每条信息中存储的内容的好信息。
为了继续我们的示例,让我们打印出有关爆炸报告中所有大于特定阈值的点击的一些摘要信息。以下代码可以做到这一点:
>>> E_VALUE_THRESH = 0.04
>>> for alignment in blast_record.alignments:
... for hsp in alignment.hsps:
... if hsp.expect < E_VALUE_THRESH:
... print("****Alignment****")
... print("sequence:", alignment.title)
... print("length:", alignment.length)
... print("e value:", hsp.expect)
... print(hsp.query[0:75] + "...")
... print(hsp.match[0:75] + "...")
... print(hsp.sbjct[0:75] + "...")
...
这将打印如下摘要报告:
****Alignment****
sequence: >gb|AF283004.1|AF283004 Arabidopsis thaliana cold acclimation protein WCOR413-like protein
alpha form mRNA, complete cds
length: 783
e value: 0.034
tacttgttgatattggatcgaacaaactggagaaccaacatgctcacgtcacttttagtcccttacatattcctc...
||||||||| | ||||||||||| || |||| || || |||||||| |||||| | | |||||||| ||| ||...
tacttgttggtgttggatcgaaccaattggaagacgaatatgctcacatcacttctcattccttacatcttcttc...
基本上,一旦您解析了RST报告中的信息,您就可以使用它做任何您想做的事情。当然,这取决于您想使用它来做什么,但希望这可以帮助您开始做您需要做的事情!
从AMPS报告中提取信息的一个重要考虑因素是存储信息的对象类型。在Biopython中,解析器返回 Record 对象, Blast 或 PSIBlast 这取决于你在分析什么。这些对象在 Bio.Blast.Record 并且非常完整。
图 表示BLAST报告的Blast Record类的类关系图。 和 PSIBBlast Record类的类图。 这是我对 Blast 和 PSIBlast 记录课程。PSIBlast记录对象类似,但支持PSIBlast迭代步骤中使用的轮。
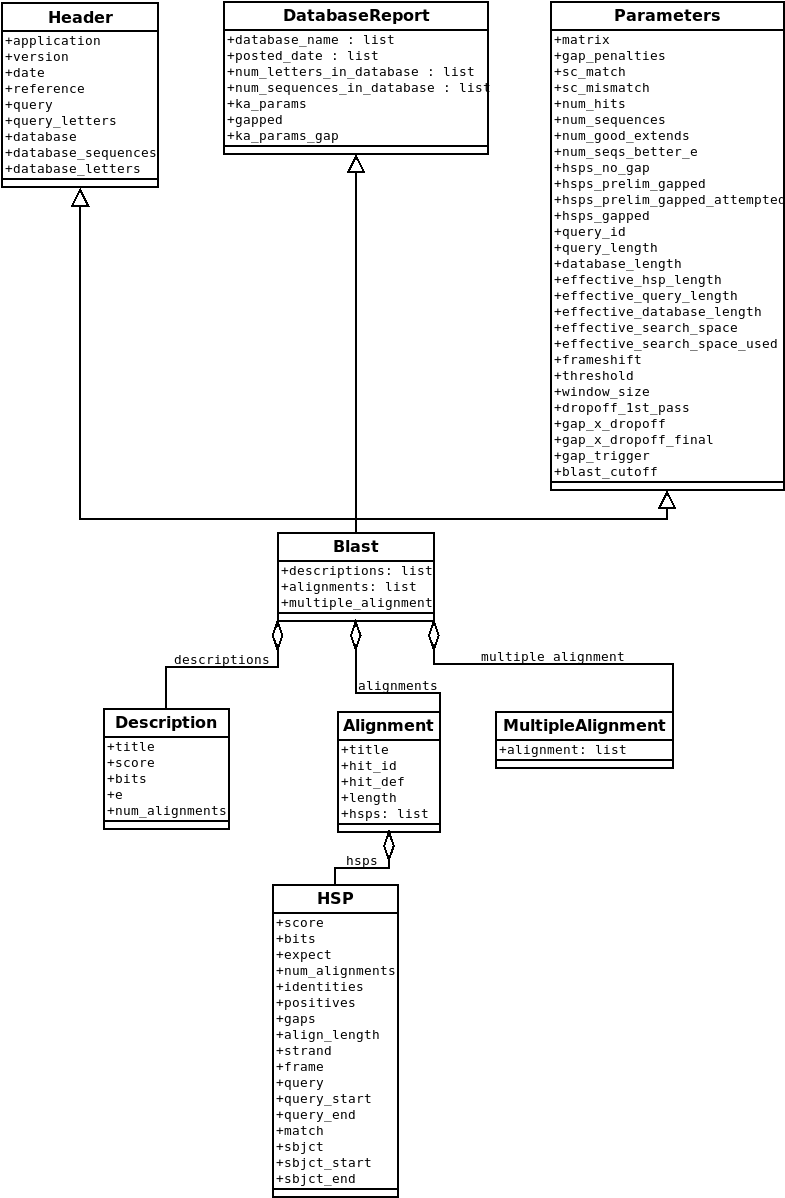
图 1 表示BLAST报告的Blast Record类的类关系图。
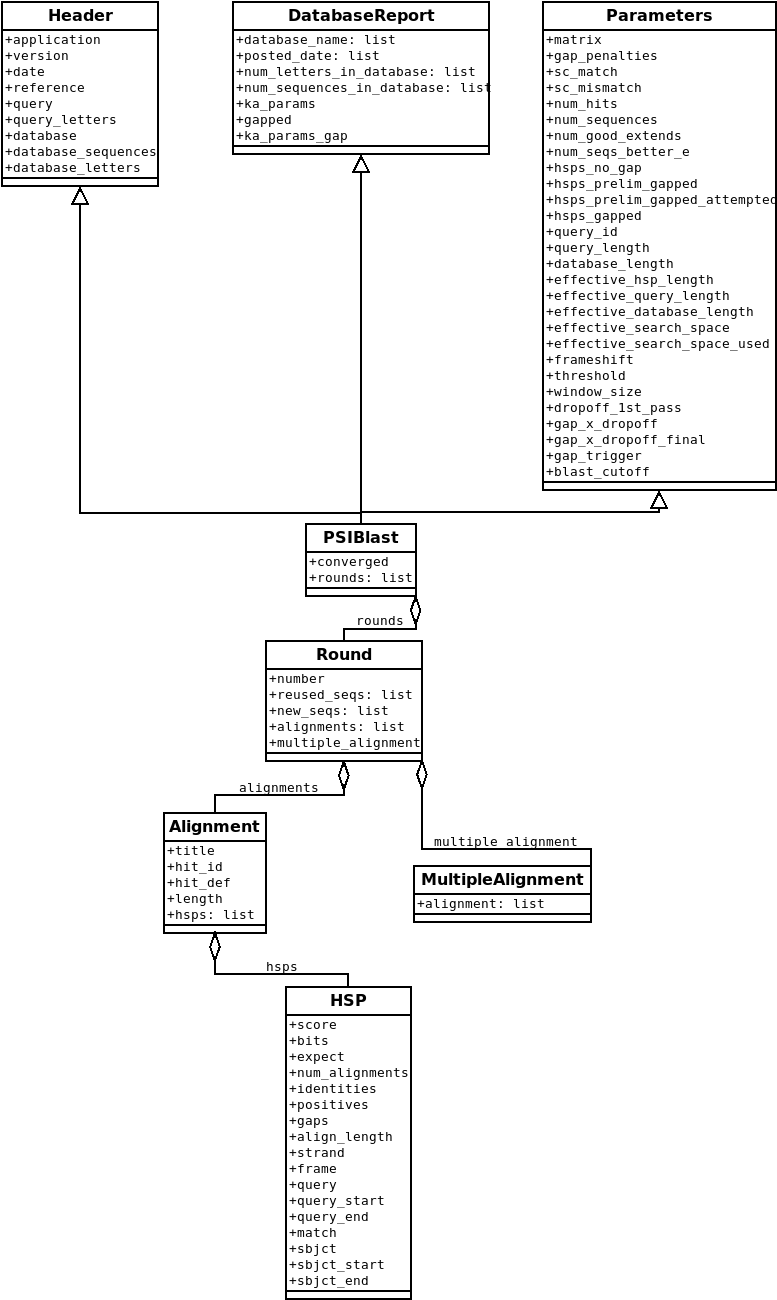
图 2 PSIBBlast Record类的类图。
如果你擅长UML,并且看到了可以做出的错误/改进,请让我知道。
处理PSI-AMPS
您可以运行PSI-BLAST(传统NCBI命令行工具)的独立版本 blastpgp ,或其替代品 psiblast )直接从命令行或使用Python的 subprocess module.
在撰写本文时,NCBI似乎不支持通过互联网运行PSI-AMPS搜索的工具。
注意到 Bio.Blast.NCBIXML 解析器可以从当前版本的PSI-BST读取ML输出,但诸如每次迭代中哪些序列是新的或重复使用的信息并不存在于该文件中。
处理RPS-AMPS
您可以运行RPS-OPS的独立版本(或者是遗留的NCBI命令行工具 rpsblast ,或同名替代品)直接从命令行或使用Python的 subprocess module.
在撰写本文时,NCBI似乎不支持通过互联网运行RPS-BLAST搜索的工具。
您可以使用 Bio.Blast.NCBIXML 解析器用于从当前版本的RPS-BST读取ML输出。